A Systematic Review on Molecular Pathophysiology Involved in Chronic Kidney Disease and the Role of Animal Models in Drug Discovery to Manage in Chronic Kidney Disease - An Update
Chronic kidney disease (CKD) is a life threatening condition that causes progressive loss of kidney function. It is one of the leading causes of death and affects about 10% of the population worldwide. CKD poses a great challenge for societies as well as health care systems. Currently, there are only a few treatments being developed for CKD, several of which have failed to improve renal outcomes. CKD is caused due to multiple factors whose mechanism is incompletely understood. Hence, various animal models have been established as an approach towards clarifying the pathogenesis and underlying mechanism of CKD. In the present review, we discuss the possible mechanisms of CKD along with the animal models used to mimic the human CKD in order to have a better understanding of generation and progression of CKD.
Abbreviations
CKD: Chronic Kidney Disease; RAAS: Renin-Angiotensin- Aldosterone System; RBF: Renal Blood Flow; DN: Diabetic Nephropathy; RAS: Renin Angiotensin System; BP: Blood Pressure; ECM: Extracellular Matrix; NSAIDs: Non-Steroidal Anti-Inflammatory Drugs; BUN: Blood Urea Nitrogen; PAN: Puromycin Aminonucleoside; SOD: Superoxide Dismutase; CAT: Catalase; GSH-Px: Glutathione Peroxidase; GFR: Glomerular Filtration Rate; ROS: Reactive Oxygen Species; NO: Nitric Oxide; UUO: Unilateral Ureteral Obstruction; NOK: Non-Obstructed Kidney.
Introduction
Chronic kidney disease (CKD) is a potentially fatal condition that affects the renal structure and function. It is characterized by progressive and irreversible loss of renal function [1, 2]. CKD is ranked 16th among the major causes of death in 2016. It is expected to rise to 5th rank by 2040. CKD affects about 10% of the world population and poses a great challenge for societies as well as health care systems worldwide [3].
In CKD, the renal damage can be ascertained when glomerular filtration rate (GFR) is <60mL/min. Based on the GFR, CKD is categorized into five stages: Stage I: Normal or high GFR (GFR>90mL/min). Stage II: Mild CKD (GFR=60-89mL/min). Stage IIIA: Moderate CKD (GFR=45-59mL/min). Stage IIIB: Moderate CKD (GFR=30-44mL/min). Stage IV: Severe CKD (GFR=15-29mL/min). Stage V: End stage (GFR<15mL/min) [4].
CKD is found to be more common in the people aged 65 years or older which accounts for 38% of cases than in people aged 45-64 years or 18-44 years which accounts for 12% and 6% cases respectively. CKD is slightly more common in women (14%) than in men (12%) [5]. There are a number of causes of CKD, out of which the most common are diabetes and hypertension. Environmental pollution, pesticides, analgesic abuse and use of unregulated food additives play a contributing role in the progression of CKD [6]. In western countries, diabetes and hypertension account for over 2/3rd of the cases of CKD. In India, diabetes and hypertension account for about 40-60% of cases of CKD. According to the data from Indian Council of Medical Research, the prevalence of diabetes has arisen to 7.1% in Indian population and it is around 28% in urban population [7]. Other possible mechanisms of CKD include haemodynamic factors, the Renin-Angiotensin-Aldosterone system (RAAS), several cytokines and growth factors, podocyte loss, dyslipidaemia and certain mechanisms of tubulointerstitial fibrosis [8].
CKD is a major contributor to health burden which is associated with high economic cost to health systems [9]. The mechanism of generation and progression of CKD is not well understood. Animal models have been extensively used as an approach towards clarifying the pathogenesis and the underlying mechanism of CKD [10]. We will be reviewing the possible mechanisms of CKD along with the animal models used to understand the generation and progression of CKD.
Pathophysiology of CKD
While discussing the mechanisms of CKD, the structural and physiological characteristics of the kidney are one of the most important aspects to be considered. In addition to these, the fundamentals of kidney injury should also be considered [11]. The renal blood flow (RBF) varies when compared with the other well perfused organs such as liver, heart and brain [11]. The RBF is about 400 mL/100 g of tissue per minute which is quite high when compared to liver, heart and brain having 100 mL, 56 mL and 80 mL per 100 g of tissue per minute respectively [12]. Due to this, the renal tissue is vulnerable to any damaging circulating agents. Also, the glomerular filtration is dependent on transglomerular pressure, which renders the glomerular capillaries susceptible to haemodynamic injury. This concludes that glomerular hypertension and hyperfiltration play a major contributing role in the progression of CKD [11]. Since nephrons are considered as the functional units of kidney, loss of these leads to non-specific wound healing processes which includes interstitial fibrosis. Infiltrating immune cells, albuminuria and, in diabetes, glucosuria, activate proximal tubular epithelial cells, which leads to secretion of proinflammatory and profibrotic mediators that promote interstitial inflammation and fibrosis [13].
There are various causes of CKD, among which the leading causes are as follows
Diabetes: Diabetes mellitus is one of the leading causes of CKD in developed countries [14]. According to the International Diabetic Federation (2015), the prevalence of diabetes was found to be 8.8% among the people aged 20 to 79 years affecting approximately 440 million people. This is expected to grow to over 550 million people by 2035 [15]. Diabetic nephropathy or Diabetic Kidney Disease is the most common complication of diabetes [16]. Diabetic nephropathy (DN) develops in about 40% of patients with diabetes and is the major cause of CKD. Diabetes leads to metabolic changes that alter the kidney haemodynamics and promotes inflammation and fibrosis [13, 17]. These metabolic changes include hyperaminoacidemia, which acts as a promoter of glomerular hyperfiltration and hyperperfusion, and hyperglycaemia [17]. The mechanism associated with glomerular hyperfiltration in diabetes is not fully understood. One proposed mechanism involves sodium-glucose cotransporter 2. Increased proximal tubular reabsorption of glucose through this cotransporter reduces the distal solute delivery, primarily sodium chloride, to the macula densa [18]. Hence, the dilation of the afferent arteriole, due to the decreased tubuloglomerular feedback, increases glomerular perfusion. The overall effect is high intraglomerular pressure and glomerular hyperfiltration [17, 18].
Hypertension: Hypertension is both cause and consequence of CKD. It is a major risk factor for both cardiovascular diseases and kidney diseases. High prevalence of hypertension is seen in patients with CKD which progresses with the severity of CKD. Various factors have been considered responsible that may cause hypertension in CKD. Some of these factors include impaired sodium excretion, activation of renin angiotensin system (RAS), sympathetic activation, imbalance in prostaglandins or kinins, etc. [19]. According to Guyton, the long term blood pressure regulation is linked to renal excretory function [19, 20]. It is assumed that renal disease intervenes with salt excretion, which leads to volume overload and resultant hypertension. This theory sheds some light on the long-term regulation of blood pressure (BP) by the kidneys. The excess salt and water retention is believed to increase the blood flow to the tissues that sets in the phenomenon of autoregulation. The tissue arterioles constrict to decrease the excessive blood flow. This resulting constriction raises the peripheral vascular resistance and hence results in hypertension [21]. Other factors such as RAS activation, sympathetic activation and imbalance of prostaglandins or kinins all cause vasoconstriction which raises the peripheral resistance and results in hypertension [19].
Renin Angiotensin Aldosterone System (RAAS): The renin angiotensin aldosterone system, also known as RAAS, plays a major role in regulating blood pressure as well as fluid balance. The RAAS is involved in the generation of a multifunctional peptide hormone Angiotensin II. Angiotensinogen is the primary substrate of RAAS which is majorly produced in liver and also in other tissues including the kidney [22].
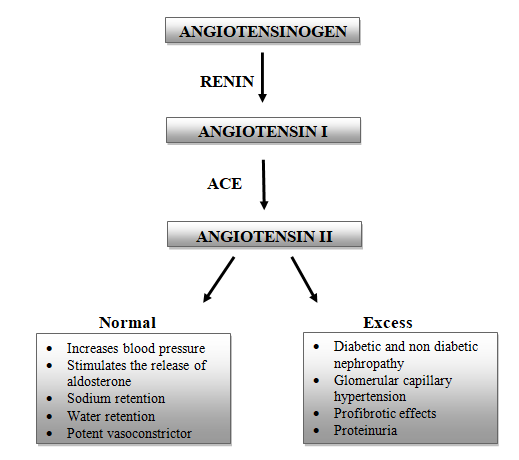
The angiotensinogen is cleaved by Renin to form Angiotensin I (Ang I) which is then subsequently cleaved to Angiotensin II (Ang II) by the enzyme Angiotensin converting enzyme (ACE). The Ang II is the major bioactive peptide generated by the RAAS [22]. Ang II causes increase in blood pressure and is also involved in sodium and water retention. Ang II also promotes secretion of Aldosterone from the adrenal gland, causing sodium retention, potassium excretion, and retention of water and also increases blood pressure [23]. Over production of Ang II and aldosterone is associated with progressive kidney damage. Diabetic and non-diabetic nephropathies, glomerular capillary hypertension, profibrotic effects and proteinuria are the clinical examples that add up to kidney injury [24].
Tubulointerstitial Fibrosis: Renal interstitium plays a central role in kidney functions and is also an important factor to be considered in the progression of CKD. The renal interstitium is the intertubular, extra glomerular and extra vascular space of the kidney. It is surrounded by tubular and vascular basement membrane and is filled with cells, extracellular matrix and interstitial fluid. Renal interstitium is said to play an important role in fluid and electrolyte exchange [25]. The interstitium accounts for about 90% of kidney volume. Tubulointerstitial fibrosis is found to be the best predictor of progression of renal disease [26]. Whenever there is a mild or limited kidney damage/injury, the tissue repair mechanisms usually repair and restore the functions. This tissue repair mechanisms comprise of a series of overlying events such as inflammation, extracellular matrix (ECM) synthesis, resolution, regeneration and remodelling. The kidneys can recover from a mild or limited damage but a persistent injury is what progresses into CKD.
Several patient renal biopsy studies and experimental models have shown that there is an overproduction of ECM, especially collagen that constitutes the scar tissue for which activation of tubule epithelial cells and interstitial fibroblasts are held responsible. The damaged epithelial cells produce collagen that manifests as thickening of basement membrane and interstitial fibrosis [27].
Podocyte Loss: Podocytes are specialised epithelial cells of the kidney that are present around the capillaries and are neighbouring cells of the Bowman’s capsule. Podocytes are considered as a major component of the urinary ultrafiltration process. These podocytes play an important role by restricting the entry of plasma protein in the urinary ultrafiltrate by forming a glomerular filtration barrier with the monolayers of fenestrated epithelial cells. This barrier consists of filtration slits between foot processes. The foot processes from one podocyte interlock with another forming a network of narrow gaps. Podocytes act by restricting the anionic and macromolecules but allow the passage of small and medium sized solutes, cationic molecules and electrolytes. When podocytes are damaged or lost, the complex structure of these podocytes is altered and this process is termed as foot process effacement. Due to this alteration, the integrity of the glomerular filtration barrier is lost [28]. Since the function of podocytes is primarily based on their complex structure, loss of these cells is highly linked to proteinuria. Proteinuria, that is the leakage of proteins into the urine, is a major indication of development of CKD [29].
Genetic and Environmental Risk Factors for CKD: Apart from the above mentioned causes, the variation in prevalence of CKD worldwide is said to be caused by the genetic or environmental or combination of both of these factors. Genetic studies have shown that genetic variants influence the development of kidney diseases. There are several examples of certain genetic variants that have high susceptibility for the development of kidney disease leading to the geographical aggregation of kidney disease. Based on the demographic data from the United States, high prevalence of CKD is seen in African American population. African Americans develop non-diabetic end stage renal disease four folds higher than Americans with European antecedentry. Another example of kidney disease found to be more common in certain groups is IgA nephropathy. People with East Asian antecedentry are predisposed to develop IgA nephropathy more frequently than others [30].
Several environmental factors have been linked to the development of CKD. These factors are implicated as the possible causes of CKD in countries or regions considered as CKD clusters where the incidence of CKD is more than average. In these clusters, it is seen that CKD is not due to the regular causes such as diabetes or hypertension but due to the influence of these environmental factors. Inspite of this suspicion, the causative role of these factors is not yet exemplified. Thus, CKD due to the unkown causes (CKDu) and infections continue to be the leading causes of CKD in most of the CKD clusters [31]. Environmental factors probably associated with development of CKD include heavy metals (like lead, mercury, cadmium, arsenic, uranium), non-steroidal anti-inflammatory drugs (NSAIDs), agricultural chemicals, industrial waste products and occupational exposures [31, 32]. Several infections have also been associated with the development of CKD such as leptospirosis, Malaria, Leprosy, Hantavirus and others [32].
Chronic Kidney Disease Models
The pathophysiology and underlying processes of kidney disease have been widely studied using animal models. The most common models used to examine the nephropathy events and treatment targets are mice and rats. These models have also been used to identify the specific disease biomarkers [10].
Adenine Induced CKD: Majority of animal models used do not truly reflect the complexities of the human condition. The rodent adenine diet model of CKD is an exception. Adding 0.75% of adenine to the diet of the rats for a period of minimum 4 weeks reflected majority of structural and functional changes observed in CKD in humans [33].
Studies reporting CKD using adenine induced model
• Yokozava T, et al. [34] reported that the prolonged feeding of adenine in rats gives a useful model for CKD studies. In this study the prolonged feeding of adenine to rats precipitated metabolic abnormalities that reflect the CKD in humans. Some of the abnormalities produced were azotemia, accumulation of uremic toxins, metabolic imbalances of amino acids and electrolytes and hormonal imbalances [34]. • Shuvy M, et al. [35] reported that due to the high adenine diet, the kidneys of the rodents were significantly enlarged due to the tubular injury. This kidney injury was linked with apoptosis along with the activation of the related pathways of apoptosis. The dietary adenine lead to a quick onset of kidney damage characterized by increased creatinine and phosphate levels, enlargement of kidneys along with damage extending about 70-80% of kidney tissue [35].
The 0.75% adenine diet model has been optimized into different dietary concentrations such as 0.075%, 0.25% and 0.5%.33 The 0.25% of adenine diet model in rats, treated for a period of 16 weeks, produced CKD having characteristics similar to CKD in humans. The damage induced was characterised by increased blood urea nitrogen (BUN), creatinine and proteinuria along with increased uric acid production [36].
Cyclosporine Induced CKD: Cyclosporine is an immunosuppressant used in transplant rejection. It is also used in other autoimmune diseases such as rheumatoid arthritis and in the treatment of graft rejection in kidney, liver and heart transplants. Cyclosporine acts as a calcineurin inhibitor that inhibits the synthesis of interleukins (ILs). These ILs are necessary for the activation and differentiation of T-lymphocytes. The difference between effective cyclosporine concentration and the concentration linked with serious toxicity is quite small. Therefore, therapeutic monitoring in patients is a necessary measure to adjust the dose to avoid rejection reactions, nephrotoxicity and other adverse effects [37]. Nephrotoxicity due to cyclosporine A (CsA) is characterized by increased resistance of afferent arteriole and efferent arteriole, reduced RBF and glomerular filtration.
Studies Reporting CKD Using Csa Induced Model
• Caires A, et al. [38] performed a study to evaluate the renoprotective effect of endothelin-1 receptor antagonists, bonestan and macitentan, in CsA induced renal dysfunction in rats. It has been reported that the group that received intra peritoneal injection of CsA (40 mg/kg) for 15 days has shown a decrease in kidney function due to increase in creatinine and urea [38]. • Venkateswarlu K, et al. [39] evaluated LOBUN, a probiotic formulation, for its nephroprotective effect on CsA induced renal failure. 20 mg/kg body weight of CsA (s.c.) was given to Wistar rats for a period of 15 days to develop renal failure. On comparision with normal group animals, control group animals had higher levels of blood parameters such as blood urea nitrogen, creatinine, uric acid and lower levels of total serum protein, indicating that the toxicity has been established in control group animals [39].
Adriamycin and Puromycin Induced CKD: The induction of nephritic syndrome by Puromycin Aminonucleoside (PAN) was first noticed in the early 1950s. A single intravenous injection of adriamycin, an anticancer antibiotic, causes a similar nephritic condition [40].
Studies reporting CKD using adriamycin and puromycin induced model
• Rider SA, et al. [41] evaluated adriamycin and puromycin for their ability to cause structural podocyte damage and increased glomerular permeability. In this study, adriamycin induced nephropathic models were well established which were characterized by the symptoms like increased protein excretion, Tubulointerstitial inflammation and fibrosis. Administration of puromycin also resulted in loss of podocytes, glomerular selectivity and terminal oedema [41]. • Munoz M, et al. [42] described the role of angiotensin II as a proinflammatory agent in adriamycin induced nephrosis in rats. This nephrotic syndrome was characterized by massive proteinuria, hypoalbuminemia, dyslipidaemia, hypercoagulability, oedema and ascites [42]. • Van der Vijgh WJ, et al. [43] conducted a study to investigate whether there is an influence on the morphometric parameters, measured in heart tissue, due to drug induced nephropathy. In this study, PAN was used to induce nephropathy in rats by administering a single injection (150 mg/kg as 2% saline solution) of PAN into the tail vein. Symptoms like proteinuria, polyuria, hypoalbuminemia, oedema and ascites represented the nephritic syndrome [43]. • Several other studies have also shown other symptoms caused by the administration of adriamycin and puromycin as a consequence of nephrotoxicity. • Bizzi A, et al. [44] reported that an extensive hyperlipemia was seen upon administration of a single dose of adriamycin (7.5 mg/kg i.v.) after 14-21 days in rats. Also, impaired apoprotein metabolism is seen as a consequence of PAN induced nephrosis [44].
Folic Acid Induced CKD: Folic acid, also known as vitamin B9, is involved in one-carbon metabolism which is essential for cellular proliferation and growth. The main sources of folic acid include egg yolk, animal livers, yeast and leafy vegetables. Deficiency of this vitamin can cause megaloblastic anaemia.45 Because of high concentration of folate receptors in the kidneys, folic acid can accumulate in greater concentration than in any other tissues. These high folate levels in the kidneys can damage cellular antioxidative processes, resulting in redox imbalance and oxidative stress. A low dose of folic acid (less than 10 mg/day ) is beneficial against oxidative stress whereas high doses (250 mg/day) are highly toxic and are used to induce kidney damage in animals.46 The pathophysiology of acute renal failure caused by the administration of folic acid is incompletely understood. Several studies have been performed in order to understand this mechanism.
Studies Reporting CKD Using Folic Acid Induced Model
• Gupta A, et al. [47] performed a study to evaluate the effect of folic acid on prooxidant state and also changes in the structure of kidney tissues. The two groups of mice, A and B, received intraperitoneal (i.p.) injection of folic acid of 100 mg/kg body weight for a period of 7 days and single dose of 250 mg/kg body weight respectively. Significant renal hypertrophy and severe renal impairment was seen in folic acid treated mice. Along with these changes, the antioxidant enzymes Superoxide Dismutase (SOD), Catalase (CAT) and glutathione peroxidase (GSH-Px) levels were markedly decreased. Therefore, the study suggests that folic acid administration resulted in oxidative stress and changes in the membrane structure that accounts for acute renal failure [47]. It has also been reported that folic acid induced nephropathy that results in acute kidney injury may progress into interstitial fibrosis. This model is the most common method used to induce patchy interstitial fibrosis [6]. • Stallions LJ, et al. [48] assessed mitochondrial dysfunction in folic acid induced acute renal injury model that rapidly developed early fibrosis. The mice were administered with a single i.p. dose of 250 mg/kg of folic acid in 300 mM NaHCO3 for 14 days. These treated mices had increased serum creatinine and urine glucose levels that ultimately decreased the glomerular function. Maximal renal dysfunction was seen after 2 days of folic acid administration, represented by elevated creatinine levels, blood urea nitrogen (BUN) and urine glucose [48].
Streptozotocin Induced Diabetic Nephropathy: Diabetic nephropathy (DN) also known as diabetic kidney disease is a leading cause of CKD and end stage renal failure worldwide. DN is represented by the structural and functional changes. It is characterized by podocyte loss, decreased endothelial cell fenestration, glomerular hyperfiltration, albuminuria, proteinuria and decreasing glomerular filtration rate (GFR) [49]. In DN, due to persistent hyperglycaemia, alterations in the redox state occur. The excessive generation of reactive oxygen species (ROS) reduces the expression of antioxidant enzymes like manganese SOD, GSH-Px and CAT [50]. Streptozotocin (STZ), also known as Streptozocin or Izostazin or Zanosar, is an anticancer agent that is categorized as antitumour antibiotic. STZ is chemically related to the drugs that are categorized as nitrosoureas used in chemotherapy [51]. STZ is diabetogenic as it is toxic to beta cells of the pancreas. STZ exerts its diabetogenic action by alkylation of DNA and also due to nitric oxide (NO) release and generation of ROS. It has also been reported that alloxan is better than STZ for animal studies since STZ has a direct effect on primary nociceptive neurons that causes painful diabetic neuropathy [52]. Several studies have been performed to evaluate the nephroprotective effect of various drugs where STZ induced DN model has been extensively used.
Studies Reporting CKD Using STZ Induced Diabetic Nephropathy Model
• Kshirsagar RP, et al. [53] evaluated the effect of geraniol on diabetes associated endothelial dysfunction in STZ treated rats. In this study, male Wistar rats were induced with diabetes by injection of STZ at 45 mg/kg intraperitoneally in 0.1 mol/L citrate buffer (pH4.5). When compared with control group animals, STZ treated rats exhibited reduced body weight, increased levels of glucose in blood and also a marked decline in plasma insulin levels. An increase in plasma level of total cholesterol, triglyceride, low and high density lipid (LDL and HDL) levels were also observed [53]. • Tzeng TF, et al. [54] evaluated the effects of Zerumbone in STZ induced DN in rats. In this study, the rats were induced with diabetes by a single intravenous injection of 60 mg/kg STZ. The STZ treated rats represented renal dysfunction due to decreased creatinine clearance, increased blood glucose levels, BUN and proteinuria, along with a significant increase in the kidney weight to body weight ratio [54].
5/6 Nephrectomy Induced CKD: 5/6 Nephrectomy (Nx) model has been extensively used in CKD researches, which is achieved by the surgical removal of 5/6 renal parenchyma [55]. This model involves the experimental reduction in renal mass by nephrectomy that leads to glomerulosclerosis and tubulointerstitial fibrosis. There are several methods to induce CKD in animals using this model. One such method is to perform uninephrectomy followed by ligation of polar branches of the renal artery. This method is frequently used in rats. Another such method involves the surgical removal of the 50% of the remaining kidney after 1-2 weeks of uninephrectomy. This approach is applicable both in rats and mice [6].
Studies Reporting CKD Using 5/6 Nephrectomy Model
• Hamzaoui M, et al. [56] performed a study that describes and compares the results of 5/6 Nx in two primary strains of mice, 129/Sv and C57BL/6JRj. Ketamine injection of 100 mg/kg (i.p.) was used to anaesthetize the mice. A left laparotomy was performed that helped in exposing the left kidney. Ligation of the upper limb of the renal artery was performed which was confirmed by the discolouration of the upper portion of the kidney. Then, a cauterizer was used to cauterize the lower pole, hilum avoided, and then sutured. A total nephrectomy (right) was performed one week post the partial Nx (left), with the exception that hilum was totally ligated and right kidney was removed. The considerable increase in levels of plasma creatinine, after 12 weeks post 5/6 Nx, established the development of CKD [56]. • Tan RZ, et al. [57] reported a new and extremely efficient 5/6 Nx model that decreased the mortality of animals. In this method, the upper pole and the lower pole of the left kidney are direct ligated after removing the right kidney one week later. As a result, the upper pole and the lower pole of this kidney undergo necrosis. This reflects the traditional 5/6 Nx. 4 and 12 weeks later, the serum creatinine, BUN and proteinuria levels were markedly elevated [57].
Unilateral Ureteral Obstruction Induced CKD: Unilateral ureteral obstruction (UUO) is used to induce kidney/renal fibrosis [58]. Renal fibrosis is the common mechanism for most of the kidney diseases [26, 58]. UUO model is consists of obstructing the urine flow which leads to tubular injury. Experimental UUO is said to reflect human chronic obstructive nephropathy. Obstructive nephropathy is a chronic clinical condition having irreversible implications which may result in Acute renal injury or chronic renal disease. The experimental procedure of UUO consists of ligating the ureter, generally using a silk thread, preferably the left kidney. The ligated kidney is referred as Obstructed Kidney (OK) and the unligated kidney is referred as Non-Obstructed Kidney (NOK). The UUO-OK exhibits characteristics such as tubular dilation, expansion of the interstitium, proximal tubular mass loss, hydronephrosis, leukocyte infiltration, and hypertrophy, death of tubular epithelial cells and presence of fibroblasts. All these modifications together result in renal tubulointerstitial fibrosis [58].
Studies Reporting CKD Using UUO Model
• Wu MJ, et al. [59] evaluated the effect of rapamycin on renal fibrosis induced by UUO. The study was performed on Sprague-Dawley rats under pentobarbital anaesthesia (i.p.). Ligation was performed on the left ureter at two points using 4-0 silk thread and cut between ligatures to avoid urinary tract infections. Layered closure of the wound was done at last. The ligation of the ureter resulted in renal fibrosis exhibited by atrophy of the tubules and deposition of interstitial matrix. Weight and length of the UUO-OK was also found to be markedly increased [59]. The UUO is generally performed on an anaesthetized animal but later on an awake model was used to prevent the anaesthetic effects. Thus, by using an anesthetized dog model it has been shown that after ureteral obstruction, there was a momentary increase in ureteral pressure and RBF followed by decrease in both the parameters [60].
Conclusion
CKD is considered a serious health condition owing to the massive rise in its prevalence and extremely huge cost of treatment [61]. CKD is defined by GFR less than 60 mL/min along with other kidney damage markers such as albuminuria, haematuria and other structural abnormalities [62]. The most prevalent causes of CKD are diabetes, hypertension and/or Glomerulonephritis. Other causes such as environmental factors like pollution, herbal medicines and pesticides are common in Asia and many developing nations [62, 63]. Early identification and staging of chronic stage of kidney disease by health care professionals are critical in decreasing the global affliction of CKD [62]. It has also been reported that an early referral to the nephrologist has shown a reduction in mortality and improved dialysis preparation [63]. Despite this, there are just few CKD treatments under development. These developing treatments shall be successful only when there is good knowledge of the pathogenesis of the disease [64].
The pathophysiology and mechanism of CKD is not well understood which can be clarified by the use of animal models of the disease [10]. Few, if any, animal models of kidney disease completely mimic all features of human pathophysiology and clinical symptoms. There are, nevertheless, numerous good models of specific characteristics of the disease [65]. These include diabetic nephropathy model, UUO model, 5/6 nephrectomy model, folic acid nephropathy model, CsA model and many more [65, 66]. The most essential learning is that no animal model can accurately mimic the human manifestations of CKD. On contrary, a well-chosen animal model can provide great insights into pathogenesis and mechanisms of CKD [66].
Acknowledgement
The authors are thankful to the Management, Principal and Staff of Acharya & BM Reddy College of Pharmacy for providing us with necessary amenities in our present study.
Author Contribution
- All authors have made substantial contributions to the the conception of the present study. The Literature search and draft was prepared by Simran Sultana.
- Dr. Uday Raj Sharma critically revised the work and approved the version to be published. All authors agree to be held accountable for the content therein.
References
-
Levey AS, Coresh J (2012) Chronic kidney disease. Lancet 379(9811): 165-180.
-
Lopez-Novoa JM, Martinez-Salgado C, Rodriguez-Pena AB, Hernandez FJ (2010) Common pathophysiological mechanisms of chronic kidney disease: therapeutic perspectives. Pharmacol Ther 128(1): 61-81.
-
Elshahat S, Cockwell P, Maxwell AP, Griffin M, O’Brien T, et al. (2020) The impact of chronic kidney disease on developed countries from a health economics perspective: A systematic scoping review. PloS One 15(3): e0230512.
-
Thomas R, Kanso A, Sedor JR (2008) Chronic kidney disease and its complications. Prim care 35(2): 329-344.
-
Centers for Disease Control and Prevention (2021) Chronic Kidney disease in the United States, 2021. Department of Health and Human Services, Atlanta, US.
-
Amarasiri SS, Attanayake AP, Jayalatika KA, Mudduwa LK (2018) Animal models of chronic kidney disease: Screening tool to investigate nephroprotective effects of natural products. Int J Pharm Chem Anal 5(2): 52-58.
-
Varma PP (2015) Prevalence of chronic kidney disease in India-Where are we heading?. Indian J Nephrol 25(3): 133-135.
-
Fogo AB (2007) Mechanisms of progression of chronic kidney disease. Pediatr Nephrol 22(12): 2011-2022.
-
Berbari AE, Daouk N, Daouk M (2018) Current Trends in Chronic Kidney Disease. J Nephrol 4(1): 7-15.
-
Bao YW, Yuan Y, Chen JH, Lin WQ (2018) Kidney disease models: tools to identify mechanisms and potential therapeutic targets. Zool Res 39(2): 72-86.
-
Matovinovic MS (2009) Pathophysiology and classification of kidney diseases. EJIFCC 20(1): 2-11.
-
Williams LR, Leggett RW (1989) Reference values for resting blood flow to organs of man. Clin Phys Physiol Meas 10(3): 187-217.
-
Romagnani P, Remuzzi G, Glassock R, Levin A, Jager KJ, et al. (2017) Chronic kidney disease. Nat Rev Dis Primers 3: 17088.
-
Winocour PH (2018) Diabetes and chronic kidney disease: an increasingly common multi‐morbid disease in need of a paradigm shift in care. Diabet Med 35(3): 300-305.
-
Sulaiman MK (2019) Diabetic nephropathy: recent advances in pathophysiology and challenges in dietary management. Diabetol Metab Syndr 11: 7.
-
Samsu N (2021) Diabetic Nephropathy: Challenges in Pathogenesis, Diagnosis, and Treatment. BioMed Res Int 2021: 1497449.
-
Alicic RZ, Rooney MT, Tuttle KR (2017) Diabetic kidney disease: challenges, progress, and possibilities. Clin J Am Soc Nephrol 12(12): 2032-2045.
-
Tuttle KR (2017) Back to the future: glomerular hyperfiltration and the diabetic kidney. Diabetes 66(1): 14-16.
-
Tedla FM, Brar A, Browne R, Brown C (2011) Hypertension in chronic kidney disease: navigating the evidence. Int J Hyp 2011: 132405.
-
Hall JE (2003) The kidney, hypertension, and obesity. Hypertension 41(3 Pt 2): 625-633.
-
Salem MM (2002) Pathophysiology of hypertension in renal failure. Sem Nephrol 22(1): 17-26.
-
Sparks MA, Crowley SD, Gurley SB, Mirotsou M, Coffman TM (2014) Classical renin-angiotensin system in kidney physiology. Compre Physiol 4(3): 1201-1228.
-
Benigni A, Cassis P, Remuzzi G (2010) Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med 2(7): 247-257.
-
Brewster UC, Perazella MA (2004) The renin- angiotensin-aldosterone system and the kidney: effects on kidney disease. Am J Med 116(4): 263-272.
-
Zeisberg M, Kalluri R (2015) Physiology of the renal interstitium. Clin J Am Soc Nephrol 10(10): 1831-1840.
-
Hewitson TD (2009) Renal tubulointerstitial fibrosis: common but never simple. Am J Physiol-Renal Physiol 296(6): F1239-F1244.
-
Hewitson TD, Holt SG, Smith ER (2017) Progression of tubulointerstitial fibrosis and the chronic kidney disease phenotype-role of risk factors and epigenetics. Front Pharmacol 8: 520.
-
Reiser J, Altintas MM (2016) Podocytes. F1000Res 5: F1000.
-
Reiser J, Sever S (2013) Podocyte biology and pathogenesis of kidney disease. Annual Rev Med 64: 357-366.
-
Friedman D, Luyckx VA (2019) Genetic and developmental factors in chronic kidney disease hotspots. Semin Nephrol 39(3): 244-255.
-
Obrador GT, Schultheiss UT, Kretzler M, Langham RG, Nangaku M, et al. (2017) Genetic and environmental risk factors for chronic kidney disease. Kidney Int Suppl 7(2): 88-106.
-
Soderland P, Lovekar S, Weiner DE, Brooks DR, Kaufman JS (2010) Chronic kidney disease associated with environmental toxins and exposures. Adv Chronic Kidney Dis 17(3): 254-264.
-
Diwan V, Brown L, Gobe GC (2018) Adenine‐induced chronic kidney disease in rats. Nephrology 23(1): 5-11.
-
Yokozawa T, Zheng PD, Oura H, Koizumi F (1986) Animal model of adenine-induced chronic renal failure in rats. Nephron 44(3): 230-234.
-
Shuvy M, Nyska A, Beeri R, Abedat S, Gal-Moscovici A, et al. (2011) Histopathology and apoptosis in an animal model of reversible renal injury. Exp Toxicol pathol 63(4): 303-306.
-
Diwan V, Mistry A, Gobe G, Brown L (2013) Adenine- induced chronic kidney and cardiovascular damage in rats. J Pharmacol Toxicol Met 68(2): 197-207.
-
Tapia C, Nessel TA, Zito PM (2018) Cyclosporine. StatPearls.
-
Caires A, Fernandes GS, Leme AM, Castino B, Pessoa EA, et al. (2017) Endothelin-1 receptor antagonists protect the kidney against the nephrotoxicity induced by cyclosporine-A in normotensive and hypertensive rats. Braz J Med Biol Res 51(2): e6373.
-
Venkateswarlu K, Heerasingh T, Babu CN, Triveni S, Manasa S, et al. (2017) Preclinical evaluation of nephroprotective potential of a probiotic formulation LOBUN on Cyclosporine-A induced renal dysfunction in Wistar rats. Braz J Pharmaceut Sci 53(2): e16041.
-
Strauch M, Gretz N (1988) Animal models to induce renal failure: a historical survey. Contrib Nephrol 60: 1-8.
-
Rider SA, Bruton FA, Collins RG, Conway BR, Mullins JJ (2018) The efficacy of puromycin and adriamycin for induction of glomerular failure in larval zebrafish validated by an assay of glomerular permeability dynamics. Zebrafish 15(3): 234-242.
-
Munoz M, Rincon J, Pedreanez A, Viera N, Hernandez- Fonseca JP, et al. (2011) Proinflammatory role of angiotensin II in a rat nephrosis model induced by adriamycin. J Renin Angiotensin Aldosterone Syst 12(4): 404-412.
-
Van der Vijgh WJ, van Velzen DI, Van der Poort SE, Schluper HM, Mross KL, et al. (1987) Morphometric study of myocardial changes during puromycin aminonucleoside induced nephropathy in rats. Anticancer Res 7(6): 1111- 1115.
-
Bizzi A, Ceriani L, Gerundino M, Spina A, Tacconi MT, et al. (1983) Adriamycin causes hyperlipemia as a consequence of nephrotoxicity. Toxicol Lett 18(3): 291- 300.
-
Lucock M (2000) Folic acid: nutritional biochemistry, molecular biology, and role in disease processes. Mol Gen Metabol 71(1-2): 121-138.
-
Yan LJ (2021) Folic acid‐induced animal model of kidney disease. Animal Models and Exp Med 4(4): 329-342.
-
Gupta A, Puri V, Sharma R, Puri S (2012) Folic acid induces acute renal failure (ARF) by enhancing renal prooxidant state. Exp Toxicol Pathol 64(3): 225-232.
-
Stallons LJ, Whitaker RM, Schnellmann RG (2014) Suppressed mitochondrial biogenesis in folic acid- induced acute kidney injury and early fibrosis. Toxicol Lett 224(3): 326-332.
-
Lim AK (2014) Diabetic nephropathy–complications and treatment. Int J Nephrol Renovas Dis 7: 361-381.
-
Miranda-Diaz AG, Pazarin-Villasenor L, Yanowsky- Escatell FG, Andrade-Sierra J (2016) Oxidative stress in diabetic nephropathy with early chronic kidney disease. J Diabet Res 2016: 7047238.
-
Akbarzadeh A, Norouzian D, Mehrabi MR, Jamshidi SH, Farhangi A, et al. (2007) Induction of diabetes by streptozotocin in rats. Ind J Clin Biochem 22(2): 60-64.
-
Rodrigues PV, Lemos BM, da Silva MV, de Campos Lima T, de Oliveira Santos D, et al. (2021) Alloxan as a better option than streptozotocin for studies involving painful diabetic neuropathy. J Pharmacol Toxicol Met 112: 107090.
-
Kshirsagar RP, Chouthe RS, Reddy GB, Kumar BD (2017) Geraniol ameliorates endothelial dysfunction in streptozotocin -induced diabetic rats. J Pharm Res 9(11): 1159-1165.
-
Tzeng TF, Liou SS, Chang CJ, Liu IM (2013) Zerumbone, a tropical ginger sesquiterpene, ameliorates streptozotocin-induced diabetic nephropathy in rats by reducing the hyperglycemia-induced inflammatory response. Nutr Metabol 10(1): 64.
-
Wang X, Chaudhry MA, Nie Y, Xie Z, Shapiro JI, et al. (2017) A mouse 5/6th nephrectomy model that induces experimental uremic cardiomyopathy. J Vis Exp 2017(129): e55825.
-
Hamzaoui M, Djerada Z, Brunel V, Mulder P, Richard V, et al. (2020) 5/6 nephrectomy induces different renal, cardiac and vascular consequences in 129/Sv and C57BL/6JRj mice. Sci Rep 10(1): 1524.
-
Tan RZ, Zhong X, Li JC, Zhang YW, Yan Y, et al. (2019) An optimized 5/6 nephrectomy mouse model based on unilateral kidney ligation and its application in renal fibrosis research. Renal failure 41(1): 555-566.
-
Martinez-Klimova E, Aparicio-Trejo OE, Tapia E, Pedraza- Chaverri J (2019) Unilateral ureteral obstruction as a model to investigate fibrosis-attenuating treatments. Biomolecules 9(4): 141.
-
Wu MJ, Wen MC, Chiu YT, Chiou YY, Shu KH, et al. (2006) Rapamycin attenuates unilateral ureteral obstruction- induced renal fibrosis. Kidney Int 69(11): 2029-2036.
-
Vaughan ED, Marion D, Poppas DP, Felsen D (2004) Pathophysiology of unilateral ureteral obstruction: studies from Charlottesville to New York. J Urol 172(6 Pt 2): 2563-2569.
-
Barsoum RS (2006) Chronic kidney disease in the developing world. N Engl J Med 354(10): 997-999.
-
Chen TK, Knicely DH, Grams ME (2019) Chronic kidney disease diagnosis and management: a review. JAMA 322(13): 1294-1304.
-
Webster AC, Nagler EV, Morton RL, Masson P (2017) Chronic kidney disease. Lancet 389(10075): 1238-1252.
-
Breyer MD, Susztak K (2016) Developing treatments for chronic kidney disease in the 21st century. Semin Nephrol 36(6): 436-447.
-
Becker GJ, Hewitson TD (2013) Animal models of chronic kidney disease: useful but not perfect. Nephrol Dialy Transpl 28(10): 2432-2438.
-
Yang HC, Zuo Y, Fogo AB (2010) Models of chronic kidney disease. Drug Discovery Today Dis Models 7(1-2): 13-19.
- Results of 6-Month Follow-Up of Patients After B-Turp and Thulep
- The Effect of Drinking Water with a High Content of Antimony and Arsenic on the Dynamics of their Distribution in the Kidneys and the Renal Excretory Function in Rats
- Effectiveness and Safety of Tansurethral Thulium Laser Enucleation of the Prostate in the Treatment of BPH: Review
- Functional Development of Kidneys in Human Ontogenesis
- Testicular Metastasis: Uncommon Prostate Cancer Case Report
- Ischemic Gangrene of the Penis: A Rare Case Report