A Comprehensive Review on HPLC Method Development, Validation, Optimization in Pharmaceuticals
The analytical method development in Pharmaceuticals is most important because it is the regulatory requirements. The determination of Active Ingredients of Pharmaceuticals (API) and Excipients is only possible when the testing method will be validated as per ICH, WHO guidelines or GMP requirements. The development will be reliable when the correct and precise strategy will be followed. Here, the systematic method development technique and validation parameters have been discussed precisely. This comprehensive review will be very helpful for analysts to develop analysis method of new molecules present in any pharmaceutical formulation. The validation concept and various techniques of validation have also been argued to create interest for the researchers.
Introduction
Analytical chemistry offers with the take a look at of the separation, quantification, and chemical identity Pharmaceutical analysis could be very crucial within the dedication of prescription drugs formula and bulk drugs regarding quality manage and high-quality guarantee. The quick increase in pharmaceutical industries of herbal and non-natural substances composed with one or more compounds or element. It is categorized into two main types, a qualitative analysis that is concerned with the identification of chemical compounds present in the sample whereas quantitative the analysis determines the number of certain elements or compounds in the sample and the producing of drugs in and around the sector cells searching for novel and advance analytical techniques in prescribed drugs. As a result, analytical method development has become a popular activity of analysis. Development in scientific and advanced analytical techniques is due to the advancement of analytical instruments. The cost and time of analysis are reduced by the improvements of the analytical method development and analytical instruments and high precision and accuracy. Techniques related to analysis are developed and validated for active ingredients, excipients, related substances, drug products, degradation products and, residual solvents, etc. for this reason become a very an important part of the requirements for regulatory agency. Analytical method development finally used in official test methods. Thus, quality control laboratories followed these methods to check the identity, efficacy, safety of products. Regulatory agencies give extreme importance in the application of analytical methods in manufacturing. Drug registration authorities demand the applicant to prove the entire process of drug development by following analytical methods. The numerous novel drugs are being manufactured and growing day by day. Therefore, it is necessary to develop novel methods and used them for controlling their quality. Advanced and developed pharmaceutical analysis needs the following requirements:” The analysis time should be minimum and economical.
- The accuracy of the analysis must comply with the guidelines of Pharmacopoeia.
- The selected method should be precise and selective. Drug analysis used for the identification, characterization, and determination of the drugs in mixtures such as in dosage forms and biological fluids. In the manufacturing process and drug development, the main purpose of analytical methods is to give information about strength, impurity bioavailability, stability, and effect of manufacturing parameters to ensure that the production of a drug product is consistent. The quality control is intended to determine and identify an exact and right product by several measures specific to avoid and get rid of errors at different stages in production. To release or discard a product is based on one or more types of control actions. Providing a simple analytical method for various complex formulations are of utmost importance. A rapid increase in pharmaceutical industries and continue production of drugs in various parts of the world have required a quick rise in and accurate new analytical a technique in pharmaceutical industries, as a result, the analytical method the development has become the basic requirements in a quality control laboratory. In this short review, the basic strategies have been discussed for method development of the drug molecules in different pharmaceutical formulations that will be more technical and informative for researchers. Why Method Development The reasons for the development of novel methods of drug analysis are: a) When there is no official drug or drug combination available in the pharmacopoeias. b) When there is no good analytical process for the existing drug in the literature due to patent regulations. c) When there are no analytical methods for the formulation of the drug due to the interference caused by the formulation excipients. d) Analytical methods for the quantitation of the analytes in biological fluids are found to be unavailable. e) The existing analytical procedures may need costly reagents and solvents. It may also involve burdensome extraction and separation procedures [1, 2, 3, 4, 5]. Method Development Strategy “Following steps are followed in the method development strategy 1. Define the application, purpose, and scope of the method 2. Define the performance parameters and acceptance criteria 3. Define validation experiments 4. Verify relevant performance characteristics of the equipment 5. Qualify materials, e.g. standards and reagents 6. Perform pre-validation experiments 7. Adjust method parameters or/and acceptance criteria if necessary 8. Perform full internal (and external) validation experiments 9. Develop SOPs for executing the method in the routine 10. Define criteria for revalidation” 11. Define the type and frequency of system suitability tests and/or analytical quality control (AQC) check for the routine. Document validation experiments and results in the validation [6].
Steps in Method Development
“In the development and manufacture of pharmaceuticals, analytical method development and validation play important roles. These methods are very helpful to ensure the identity, purity, potency, and efficacy of drug products. During method development many factors to be considered. Initially, physiochemical properties (pKa, log P, solubility) and mode of detection (suitable wavelength in case of UV detection) [7] might be appropriate for evaluation. The principle reason of the HPLC approach is an effort to the separate amount of the principle active drug, any interfering impurities, all available artificial intermediates, and any degradants. Steps to be followed in approach improvement are: Recognize the physicochemical homes of drug molecule
- Set up HPLC situations
- Instruction of pattern answer for technique improvement
- Approach optimization
- Validation of approach (eight)”
Physicochemical Properties of a Drug Molecule
Physiochemical properties of a drug molecule are very important in method development. For method development, it is necessary to study the physical properties like pH, pKa, polarity, and solubility of the drug molecule. Polarity: The Polarity of a compound helps an analyst, to decide the solvent composition of the mobile phase. In a nonpolar covalent bond, the electrons are equally shared between two atoms but a polar covalent bond are one in which one atom has a greater tendency to attract the shared electrons than other atoms [8]. Solubility: The solubility is very helpful to explain the polarity of molecules. Polar, e.g. water, and nonpolar e.g. benzene, solvents do not mix. In general, like dissolves like i.e. materials with the same polarity are soluble in each other. The choice of diluents is selected on the basis of solubility of analytes. The analytes must be soluble in the diluents and must not have an affinity to react with any other components.
PH and PKA: The pH and PKa plays an important role in HPLC method development. The acidity or basicity of a substance is decided typically by the pH value. A suitable pH selection for ionizable analytes often gives symmetrical and sharp peaks because symmetrical and sharp peaks are necessary in quantitative analysis in order to get low detection limit, low relative standard deviations between injections, and reproducible retention times. The pKa is characteristic of a particular compound, and it expresses how readily the compound provides up a proton [9].”
Set Up HPLC Conditions
“A buffer is a partially neutralized acid which inhibits changes in pH. Buffering capacity is the ability of the buffer to inhibit changes in pH (i). Buffer capacity rises as the molar concentration of the buffer salt/ acid increases (ii). The closure of the buffered pH to the pKa, more the buffer capacity (iii). Buffer capacity is explained as the molarity of Sodium Hydroxide required to increase the pH by 1.0. The effect of pH on analyte retention, type of buffer selected, solubility in organic modifier and its effect on detection is significant in reversed-phase chromatography (RPC) method development of ionic compounds. An inappropriate buffer selection, in terms of buffer species, ionic strength, and pH can result in poor or non-reproducible retention and tailing in reverse phase separation of polar and ionizable compounds [9].
Important information concerning sample composition and properties [4].
- Number of compounds present
- Chemical structures (functionality) of compounds
- Molecular weights of compounds Pka value of compounds
- UV spectra of compounds
- The concentration range of compounds in sample of interest
- Sample solubility”
Buffers Selection
“Obtaining exact and sharp peaks for analytes under interest needs a proper buffer of a mobile phase. Buffer improves the peak sharpness of the compounds and helps to change the selectivity and retention of acidic or basic compounds. The recommended concentration of 10-20mM of ammonium salts of your choice as a starting buffer (i.e. acetate, formate, carbonate, or phosphate salts) for electrolyte solutes. Buffers are the solution of a weak acid and its conjugate base, or a weak base and its conjugate acid. They mitigate the effect of hydrogen/ hydronium and hydroxide ions as results reduce the changes, even upon dilution. The characteristic pH the range for reverse-phase on silica-based is usually pH 2.0 to 8.0. It is compulsory that the buffer has a pKa near to the required pH. Subsequently buffers control pH best at their pKa. A rule of thumb is to follow a buffer with a pKa value [10] (Table 1).”
| PKa (at 25℃) | Compound |
|---|---|
| 0.3 | Trifluoroacetic acid2 |
| 2.15 | Phosphoric acid (pk1) |
| 3.13 | Citric acid (pk1) |
| 3.75 | Formic acid |
| 4.75 | Acetic acid |
| 4.76 | Citric acid (pk2) |
| 4.86 | Propionic acid |
| 6.35 | Carbonic acid (pk1) |
| 6.4 | Citric acid (pk3) |
| 7.2 | Phosphoric acid (pk2) |
| 8.06 | Tris |
| 9.23 | Boric acid |
| 9.25 | Ammonia |
| 9.78 | Glycine (pk2) |
| 10.33 | Carbonic acid (pk2) |
| 10.72 | Triethylamine |
| 11.27 | Pyrrolidine3 |
| 12.33 | Phosphoric acid (pk3) |
Table 1: Merck Index and CRC Handbook of Chemistry and Physics [11].
Mobile Phase Selection
Selecting the accurate composition and type of mobile is important because it is adjustable that governs separation. However, the choice is restricted because of the stationary phase employed in the column used. The clear difference is between reversed-phase and normal chromatography. In normal phase system, nonpolar solvents such as hexane or iso-octane while in reversed-phase chromatography polar solvents such as water acetonitrile or methanol are used. The selection of the mobile phase is governed by the physical parameters of the solvent. Polarity, miscibility with other solvents, chemical inertness, UV cut off wavelength and toxicity are factors to be considered in mobile phase selection [12] (Table 2).”
| A-Normal phase | ||
|---|---|---|
| Solvent | Polarity index | UV cutoff (nm) |
| Hexane | 0.1 | 210 |
| Isooctane | 0.1 | 205 |
| Diethyl ether | 2.8 | 218 |
| Dichloromethane | 3.1 | 205 |
| B- Reverse phase | ||
| Water | 10.2 | 200 |
| Methanol | 5.1 | 210 |
| Acetonitrile | 8.8 | 210 |
| Tetrahydrofuran | 4 | 280” |
Table 2: Typical solvents for HPLC mobile phases [10].
Preparation of Sample Solutions for Method Development
The drug active compound being analyzed should be stable in a diluent. In the initial stage of method development, the diluent should be kept in the amber color flask until it is determined that active substance is stable at room temperature and does not affect under normal laboratory conditions. The solution should be filtered through 0.22 or 0.45µm pore-size filter paper is generally preferred for the removal of particulates. Filtration is a precautionary maintenance tool for analyses [13, 14]. Sample preparation is an important step in method development that the analyst must study. The efficiency of the syringe filters is mostly determined by their ability to eliminate containments without leaching unwanted extractable into the filtrate. If undesired peaks are seen in the filtered samples, then the diluent (solvent) must be filtered to remove leachable component comping from the syringe filter housing or filter [9] (Figure 1).
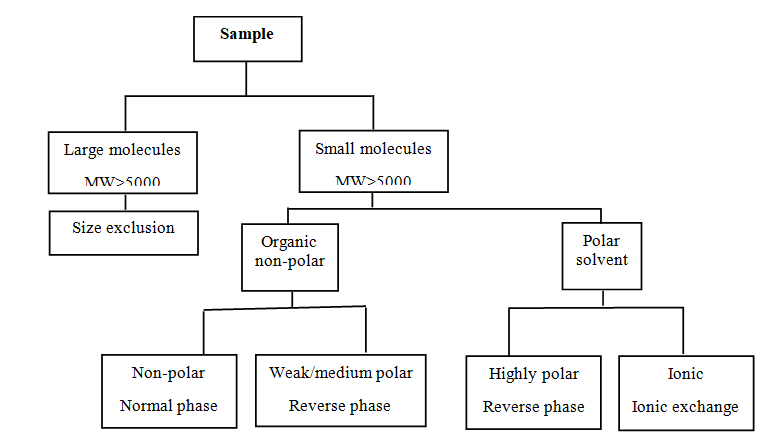
HPLC method development mainly focused on the optimization of HPLC conditions [15]. The mobile phase and stationary phase compositions should be considered. Optimization of mobile phase parameters is always to be taken into account first as this is much easier and convenient than stationary phase optimization. To reduce the number of trial chromatogram involved, only the parameters that are likely to have an important effect on selectivity in the
Validation
Although validation studies have been carried out in the pharmaceuticals industries for a long time, the validation interest is growing in recent years on quality assurance programs and is important to an efficient manufacturing process. Validation concept that has grown in the United States in 1987. The concept of validation has followed through the years to adopt a wider range of activities from analytical methods used for the quality control of drug ingredients and drug products to computerized systems for clinical trials, process control or labeling. Validation is a vital and an important part of cGMP. The word validation means in simple words assessment of validity, or action of assuring effectiveness. Validation is teamwork involving people from different departments of the industry. Method validation is the process of “designing documented evidence” which assures a high degree of assurance that product (equipment) will comply with the requirements for the intended analytical applications [6].” Importance of Validation
- Assurance of quality
- Time-bound
- Process optimization
- Reduction of quality cost
- Nominal mix-ups, and bottlenecks
- Minimal batch failures improved efficiency and productivity
- Reduction in rejections
- Increased output
- Avoidance of capital expenditures
- Fewer complaints about the process related failures.
- Reduced testing in process and in finished goods
- More rapid and reliable start-up of new equipment
- Easier scale-up form development work
- Easier maintenance of equipment
- Improved employee awareness of processes
- More rapid automation.” Types of validation There are main four types of validation used in pharmaceuticals industries
- Process validation
- Analytical method validation
- Equipment validation (DQ, IQ, OQ, PQ)
- Cleaning validation Process Validation: Process validation” is developing documented evidence which gives a high degree of assurance that specific processes consistently produce product conforming its predetermined specifications and quality attributes [16, 17].
Analytical Method Validation: There are many reasons for the requirement to validate analytical procedures. Among them are good science, regulatory requirements and quality control requirements. The Code of Federal Regulations (CRF) 311.165c explicitly describes that “the accuracy, sensitivity, and reproducibility of test methods used for firm shall be developed and documented. Finally, the management of the quality control unit would definitely want to ensure that the analytical methods that the department follows to market its products are properly validated for intended use so the product will be harmless for human use [18, 19].” The process explains a framework for validation of pharmaceuticals methods. Results from the validated method can be considered to check its quality, reliability as well as consistency regarding analytical results. In pharmaceutical industries method validation is a crucial part of the quality- control system and of validation of an analytical method is used to confirm that applied procedures for a specific test comply with the intended requirements. Different Guidelines like USP, ICH, and FDA, etc. can secondary regulation of good manufacturing practices unavoidably needs assay validation [20, 21, 22, 23, 24].”” Equipment Validation: “Equipment validation is a part of Quality Assurance process of establishing a document that a piece of equipment satisfies its intended requirements.
Cleaning Validation: It is the methodology used to check the cleaning process that removes chemical and microbial residues of the active, excipients ingredients of the products manufactured in equipment [25].
Types of analytical procedures The four most common types of analytical procedures are:
- Identification tests
- Quantitative tests for impurities content
- Limit tests for the control of impurities
- Quantitative tests of the active moiety in samples of a drug Identification tests are used to ensure the identity of active ingredients in the sample.
This generally obtained by comparison of a property of the sample (e.g., spectrum, chromatographic behavior, chemical nature, etc.) to that of a reference standard. Different validation parameters are required for a quantitative test than a for a limit test. Assay methods are used to quantify the analyst present in a given sample [26].” Develop a validation protocol or operating procedure for the Validation.
Strategy for the Validation of Methods
Method development and validation are an iterative process. The influence of operating parameters on the performance of the method can be assessed at the validation of a stage that was not done during the development/ optimization stage of the method. The most significant point raised for validation is that the validity of a method can be demonstrated only through laboratory studies. It is not sufficient to simply review historical results; instead, laboratory studies must be conducted which are intended to validate the specific method, and those studies should be pre-planned and described in suitable documentation. This documentation should clearly indicate the method’s intended use and principles of operation, as well as the validation parameters to be studied, and a rationale for why this method and these parameters were chosen. It also must include pre-defined acceptance criteria and a description of the analytical procedure.”
Parameters for Method Validation
The parameters for method validation are:
Accuracy
The accuracy is the degree of closeness of measurements of a quantity to that of true value [27].
In a method with high accuracy, a sample (whose “true value” is confirmed) is checked and the measured value is the same as the true value. There are three methods to determine accuracy:
- Comparison to a reference standard
- Recovery of the analytes spiked into a blank matrix
- Standard addition of the analytes It should be known how the individual or total impurities are to be determined [5].
Precision (Repeatability and Reproducibility)
The precision of measurements, involved in reproducibility and reproducibility, is the degree to which repeated values under constant conditions show the same results [28]. The precision of the analytical method explains the closeness of agreement between a series of values obtained from multiple sampling of the same homogenous sample under given conditions. Precision can be studied at three levels: repeatable, intermediate precision and reproducibility. Precision should be studied using homogeneous, true samples. The precision of an analytical method is usually expressed as the standard deviation, variance or coefficient of variation of a series of values [29].
Linearity and Range
The linearity of a method is a measure that tells us how a calibration plot of response versus concentration approximates a straight line. The data is then represented using linear least-square regression. The obtained plot slope, intercept and correlation coefficient provide the desired information on linearity [5].
Linearity describes that obtained results in the experiment are directly proportional to the concentration of analytes in the sample [30]. The linearity of the analytical method expresses that the obtained results are directly proportional to the concentration of analytes in the sample but the range is the smallest and largest concentration that sets a linear relationship between concentrations and the response of the method. The ICH guidelines do not demand any proof of precision, though it is clear that without satisfactory precision, the linear relationship cannot be guaranteed. Mostly, linearity is demonstrated by the least square regression. Sometimes it important to transform the data to attain a linear fit [31]. Linearity is determined by replicate injections of 5 or more concentrations level within the range of 40–160 %. The response should be directly proportional to the concentrations of the analytes or proportional by means of a well-defined mathematical calculation. Linearity is evaluated graphically by plotting a graph of the relative responses on the y-axis and the corresponding concentrations on the x-axis. A linear regression equation is applied to the results to evaluate the correlation coefficient. In addition, y-intercept, the slope of the regression line and residual sum of squares should also calculate. The range of an analytical method is the interval between the upper and lower concentrations (amounts) of analytes in the sample (including these concentrations) for which it has been demonstrated that the analytical procedure has a suitable level of precision, accuracy, and linearity. The range is normally expressed in the same units as the test results (e.g., percentage, parts per million) obtained by the analytical method [32].”
Limit of Detection (LOD) and Limit of Quantitation (LOQ)
“The detection limit of an analytical method is the lowest amount of analytes in a sample that can be detected but not necessarily quantitated as an exact value. In chromatography, the detection limit is the injected amount that results in a peak with a height of at least two or three times as high as the baseline noise level. Besides this signal/noise method, LOD can be measured by another three different methods; (i) visual inspection (ii) standard deviation of the blank response (iii) standard deviation of the response based on the slope of the calibration curve. The quantitation limit of an analytical method is the lowest amount of analytes in a sample that can be quantitated with suitable precision and accuracy. In chromatography, the quantitation limit is the minimum injected an amount that produces quantitative measurements in the target matrix with acceptable precision, typically requiring peak heights 10 to 20 times higher than the baseline noise. Beside this signal/noise method, LOQ can be measured by another three different methods; (i) visual inspection (ii) standard deviation of the response (iii) standard deviation of the response based on the slope of the calibration curve [32].
Selectivity/ Specificity
The specificity of an analytical procedure is to measure the concentration of an analyte in the presence in the presence of all potential sample active materials and excipients such as the starting materials, intermediates in the synthesis, and inactive ingredients in the final products, and the degradation of products.
Specificity in liquid chromatography is carried out by selecting optimal columns and setting chromatographic conditions, such as mobile phase composition, column temperature and detector wavelength. The sample preparation procedure is also optimized for the best separation. Specificity can be expressed by analyzing the sample containing impurities or other materials spiked onto analytes of interest. The degradation of products could be produced by storing the analytes under stress conditions. Usually, stress conditions for the production of degradation products for active pharmaceutical ingredients are heated (500C, 600C), 6500 lx UV light), acidic condition (in 0.1mol/L HCl solution), and oxidant (3% H2O2 solution). Heat, light, and humidity are the main factors that are responsible for severe conditions. Resulting mixtures samples should be checked for analysis, and the peak purity and resolution of analytes is evaluated from the nearest eluting peak. In chromatographic analyses, it is difficult to estimate whether the peaks in the chromatogram are pure, or consists of more than one compound. In the past, chromatographic parameters such as mobile phase composition were changed in order to determine the peak purity. Recently VV/VIS diode-array detectors are being used [33].” Robustness & Ruggedness “The robustness of the analytical procedure is a parameter of method validation its capacity to remain unchanged by a small, but a deliberate change in method parameter and provides an indication of its reliability during normal usage by changing, 1. Change in pH of mobile phase (±0.2) 2. Influence of change in flow rates (±10%). Ruggedness (system to system variation), Ruggedness of the analytical method is the degree of reproducibility of test results gained by the analysis of the same sample under different conditions such as laboratory, analyst and instruments [34].
System Suitability Studies
System suitability tests are a very important part of HPLC methods and used to check that the accuracy and precision of the system are suitable for the analysis to be determined [35]. System suitability is explained as the checking of a system before or during analysis to ensure system performance. Minimum five injections of the sample (50µg/ml) were utilized for calculating system suitability parameters like % RSD, Tailing factor, and theoretical plates [34].
The parameters used in the system suitability tests (SST) report is as follows:”
- The number of theoretical plates or Efficiency (N).
- Capacity factor (K).
- Separation or Relative retention (α).
- Resolution (Rs).
- Tailing factor (T).
- Relative Standard Deviation (RSD).
GMP Requirements
The pharmaceutical analysis may be defined as that branch of practical chemistry which deals with the resolution, separation, identification, determination, and purification of a given sample of a medicine or a pharmaceutical product; the detection and estimation of impurities that may be present are also included. The quality standard of a pharmaceutical product is monitored and maintained throughout the process of manufacture and also in the finished product by a series of quality control tests [36].
ICH and WHO guidelines suggest that the developed analytical method of pharmaceuticals finished products should be precise, accurate, stable and validated. In this research work ICH and WHO guidelines were, therefore, followed for the method validation [37].
Validation is the procedure of documentary proof of method, process, or activity followed in the testing method and then maintains the stated level of compliance at all stages. Analytical method validation in the pharmaceutical industry is very important for the final testing and compliance of products, it also provides the confidence the procedure will consistently produce the intended results [38].
HPLC method validation is the technique used to check that the HPLC procedure used for a specific testing method for the intended use. The results obtained from method validation tell us the reliability and reproducibility of a procedure used. Method validation has much value in literature and from the industrial committee and regulatory agencies. The International Conference on Harmonization (ICH) of Technical Requirements for the Registration of Pharmaceuticals for Human Use [22] has established a harmony text on the validation of analytical procedure. The documents contain definitions for eight validation parameters. ICH also made appendices with extra detail methodology [22].
The United States Food and Drug Administration (US FDA) has recommended guidelines on submitting samples and analytical facts for methods validation [39]. The United State pharmacopeia (USP) has issued explicit guidelines for method validation for compound estimation [40].
The USA FDA has supplemented a section 211.222 on ‘method validation, to cGMP regulations [41]. This suggests the manufactures to create and document the accuracy, sensitivity, reproducibility and other parameters necessary to validate method validation. The validation is also needed to comply with the existing requirements for laboratory archives given at sec.211.194 (a). These requirements consist of a statement of each method applied in testing the sample to comply with appropriate standards of accuracy and reliability as used to the tested product.”
Conclusion
This article provides a thought that what is validation, its type, why it is essential, how to develop a method and how to carry out the validation procedure to reveal that the technique is able for its proposed reason. All validation parameters such as LOD, LOQ, Linearity, Range, selectivity, specificity, robustness, ruggedness, accuracy and precision are explained. In this review, different types of validation have also been discussed that are conducted at various processes or stages of processes of pharmaceutical industry. Because validated process or method is necessary to manufacture a safe and quality product.
References
-
Lal B, Kapoor D, Jaimini M (2019) A review on analytical method validation and its regulatory perspectives. Journal of Drug Delivery and Therapeutics 9(2): 501- 566.
-
Gassmann O, Reepmeyer G, Zedtwitz M (2008) Trends and Drivers for Growth in the Pharmaceutical Industry. 2nd (Edn.), Leading Pharmaceutical Innovation. Springer Berlin, Heidelberg, pp: 186.
-
Breaux J, Jones K, Boulas P (2003) Understanding and Implementing Efficient Analytical methods development and validation. Pharmaceutical online, pp: 6-13.
-
Snyder LR, Kirkland JJ, Glajch JL (1997) Practical HPLC method development. 2nd(Edn.), Wiley, pp: 800.
-
Ravisankar P, Navya CN, Pravallika D, Navya Sri D (2015) A review on step by step analytical method validation. IOSR Journal of Pharmacy 5(10): 7-19.
-
Tangri P, Rawat PS (2012) Validation A critical parameter for quality control of pharmaceuticals. Journal of Drug Delivery and Therapeutics 2(3).
-
Chandrul Kaushal K, Srivastava B (2010) A process of method development: A chromatographic approach. Journal of Chemical and Pharmaceutical Research 2(2): 519-545.
-
Riddhiben MP, Piyushbhai MP, Natubhai MP (2011) Stability indicating HPLC method development a review. International Research Journal of Pharmacy 2(5): 79-87.
-
Gupta V, Jain ADK, Gill N, Gupta K (2012) Development and validation of HPLC method a review. International Research Journal of Pharmaceutical and Applied Sciences 2(4): 17-25.
-
Patwekar S, Sakhare RS, Nilesh NN (2015) HPLC method development and validation A general concept. International Journal of Chemical and Pharmaceutical Sciences 6(1): 8-14.
-
Dolan J (2016) A guide to HPLC and LC-MS buffer selection. ACE HPLC Columns–ultra inert base- deactivated HPLC columns, pp: 1-20.
-
Sabir AM, Moloy M, Bhasin PS (2013) HPLC method development and validation A review. International Research Journal of Pharmacy 4(4): 39-46.
-
Mayer ML (1996) Selecting filters for chromatographic applications. LC GC 14(10): 902-905.
-
Mayer M (1997) Filtration Preventative maintenance for HPLC. American laboratory Fairfield 29(1): 34-37.
-
Kardani K, Gurav N, Solanki B, Patel P, Patel B (2013) RP HPLC method development and validation of gallic acid in polyherbal tablet formulation. Journal of Applied Pharmaceutical Science 3(5): 37-42.
-
Nash RA, Wachter AH, Swarbrick J (2003) Pharmaceutical process validation: an International 3rd Edition Revised & Expanded. In: Berry IR, et al. (Eds.), Drug and the pharmaceutical sciences. Taylor 129: 1-883.
-
Lavanya G, Sunil M, Eswarudu MM, Eswaraiah MC, Harisudha K, et al. (2013) Analytical method validation An updated review. International Journal of Pharmaceutical Sciences and Research 4(4): 1280-1286.
-
Ravisankar P, Rajyalakshmi G, Devadasu Ch, Rao GD (2014) Instant tips for right and effective approach to solve HPLC trouble shooting. Journal of chemical and pharmaceutical sciences 7(3): 259-274.
-
Agalloco J (1995) Validation an unconventional review and reinvention. PDA Journal of pharmaceutical science and technology 49(4): 175-179.
-
Carr GP, Wahlich JC (1990) A practical approach to method validation in pharmaceutical analysis. Journal of pharmaceutical and biomedical analysis 8(8-12): 613- 618.
-
Swartz ME, Krull IS (2012) Handbook of analytical validation. 1st(Edn.) Routledge taylor & francis group.
-
International Conference on Harmonization ICH of Technical Requirements for Registration of Pharmaceuticals for Human Use (2005) ICH Harmonised Tripartite Guideline Quality Risk Management Q9, pp: 1-23.
-
FDA U, FDA (2000) Guidance for Industry. Analytical Procedures and Method Validation, pp: 1-37.
-
Orr JD, Krull IS, Swartz ME (2003) Validation of impurity methods Part II. LC GC North America. Intellisphere LLC 21(12): 1146-1181.
-
Tanyous JN (2019) Cleaning Validation Complete Guide for Health-Based Approach in Chemical Cross- Contamination Risk Assessment. PDA journal of pharmaceutical science and technology 73(2): 204-210.
-
Ravichandran V, Shalini S, Sundram K, Harish R (2010) Validation of analytical methods strategies importance. International Journal of Pharmacy and Pharmaceutical Sciences 2(3): 18-22.
-
Bièvre PD (2009) The 2007 International Vocabulary of Metrology VIM JCGM 200 2008 ISO/IEC Guide 99 Meeting the need for intercontinentally understood concepts and their associated intercontinentally agreed terms. Clinical biochemistry 42(4-5): 246-248.
-
Taylor J (1997) Introduction to error analysis. 2nd(Edn.), the study of uncertainties in physical measurements, pp: 1-343.
-
Guideline IHT (2005) Validation of analytical procedures text and methodology Q2 R1. International conference on harmonization, pp: 1-15.
-
Kruve A, Rebane R, Kipper K, Oldekop ML, Evard H, et al. (2015) Tutorial review on validation of liquid chromatography mass spectrometry methods Part I. Analytica chimica acta 870: 29-44.
-
Walfish S (2006) Analytical methods a statistical perspective on the ICH Q2A and Q2B guidelines for validation of analytical methods. BioPharm International 19(12): 40-45.
-
ICH M (1996) Q2B validation of analytical procedures methodology. proceeding of the International conference on Harmonization.
-
Kazusaki M, Ueda S, Takeuchi N, Ohgami Y (2012) Validation of analytical procedures by high performance liquid chromatography for pharmaceutical analysis. Chromatography 33(2): 65-73.
-
Kumar S, Jamadar LD, Bhat K, Musmade PB, Vasantharaju S, et al. (2010) Analytical method development and validation for aspirin. International Journal of Chem Tech Research 2(1): 389-399.
-
Shabir GA (2004) A practical approach to validation of HPLC methods under current good manufacturing practices. Journal of validation technology 10: 210-218.
-
Kealey D, Haines P (2002) Analytical Chemistry instant notes. 1st (Edn.), Bios Scientific Publishers.
-
Desai KB, Patel MA, Pradhan PK, Dey S, Upadhyay UM (2013) Simultaneous estimation of sparfloxacin and dexamethasone in bulk and in their combined dosage form by HpLc method. Asian Journal of Pharmaceutical Research and Development 1(5): 55-62.
-
Laghave PK, Gite SV (2017) An Overview on Validation Technique. Journal of Current Pharma Research 8(1): 2249-2254.
-
FDAU (1987) Guidelines for submitting samples and analytical data for methods validation. Center for Drugs and Biologies, Department of Health and Human Services, Rockville, MD.
-
Chapter G (1995) 1225>, Validation of compendial methods. United States Pharmacopeia XXIII, National Formulary, XVIII, Rockville, MD, the United States Pharmacopeial Convention.
-
CFR (1978) Part 211: Current Good Manufacturing Practice for Finished Pharmaceuticals.
- Electronic Waste Management in the Top Ten Economies in the World: A Critical Review on Waste Generation, Regulations, Collection, Recycling and Environmental Challenges
- Some Challenging Transdisciplinary Aspects of the Sustainable Waste Management in the Permacrisis Context
- Use of Mobile Autonomous Systems for Pollution Control of Inland Water Bodies
- Environmental Impact Perspective Sustainable Online Textile Retailing: Harnessing Augmented Reality-Based Digital Twins in Bangladesh
- Composite Treatment Module for Removing Acidity and Metal (Loid)S from Acid Rock Drainage
- Household E-Waste Management Systems [E-Wms] in Malaysia