Autosomal Dominant Polycystic Kidney Disease: At a Glance
<p>Polycystic kidney disease (PKD) is a systemic disorder which adds majority of renal patients to end stage renal disease (ESRD). Among all type of PKD, autosomal dominant polycystic kidney disease (ADPKD) is most prevalent and fourth most leading cause of dialysis and kidney transplant. Polycystic kidneys in several genetic and non-genetic disorders suggest the role of multiple factors in cystogenesis. Linkage analysis revealed some closely linked loci two of which are identified as PKD1, PKD2 and an unidentified locus to ADPKD. Candidate gene products are central molecules of proteinprotein complex located at primary cilia and adhesion junctions of cell. In present study, Adult age group 31-59 years patients were more in number and relatively enlarged kidney size was observed in 49 PKD cases. Almost equal incidence of PKD in females to males (48.8%:51.2%) was observed in Indian population. 30.95% of the total cases enrolled cases had positive family history. Two associated symptoms like hypertension and presence of liver cysts and hepatomegaly were found more in frequency 19% and 39% in polycystic patients. Expressivity and variability in symptoms of ADPKD at individual level imply the role of genetic heterogeneity, effect of modifier genes and environmental factors.</p>
Introduction
Polycystic kidney is most frequently found renal abnormality associated with several genetic and non- genetic disorders. It is a condition in which unilateral or bilateral presence of multiple cysts; disrupt the precise architecture of kidneys [1, 2]. Among all types of PKD, worldwide prevalence of ADPKD is 1:1000 and contributes 10% of ESRD in humans [2, 3]. ADPKD primarily found in adults and are equally frequent in men and women in western world [4]. Linkage study showed the involvement of two known loci PKD1 and PKD2 to the ADPKD [5, 6, 7, 8, 9]. Some affected families did not shown linkage to either PKD1 or PKD2 suggesting the involvement of unidentified gene/s [10, 11, 12, 13].
Clinical description
Biochemical examinations of patients revealed increased level of blood urea and serum creatinine [14]. Cardinal symptoms of ADPKD are adult onset of bilateral, multiple renal cysts of variable sizes originated from 1- 3% of all nephrons. Patients have complications of flank pain, hematuria, frequent urinary tract infection, cyst infection and cyst hemorrhage. Sometimes renal stone, extra renal cyst such as liver cyst, pancreatic cyst, cyst in testis and ovary are also present in ADPKD patients. Non- cystic manifestations like cardiovascular defects (25%), intracranial aneurysm (8%) and hypertension (70-80%) are also common in these patients [15]. There are some adult and pediatric cases found in Indian and other population [16] which state the concurrence of ADPKD with other pathophysiological conditions like diffuse proliferative glomerulonephritis [17, 18], situs inversus [19], polycystic liver disease [20], cystic hemorrhage in kidney [21], recurrent pneumothorax [22]. Other gastrointestinal and musculoskeletal complications like diverticulosis and hernia are also observed in patients [23]. Genetic screening of affected individuals revealed the two genes and polycystic kidneys are result of genetic insult. Affected individuals with PKD1 defect manifest the disease condition at third to fourth decade of life and most of them reached to ESRD by the age of 55 years. However, age of disease onset for PKD2 defect is fourth to fifth decade of life and reaches to ESRD by the age of 70 years [24]. ADPKD has been reported in both familial and sporadic cases suggesting that both germinal and somatic mutations might be involved in cyst generation [25]. Based on clinical symptoms, radiology techniques help a lot to examine and collect the status of kidneys. Ultrasonography, CT scan and MRI are helpful in suspected individual’s clinical diagnosis because small sizes cysts in kidneys. Ravine has defined the criteria to classify the ADPKD based on non-invasive techniques [26].
Genetics
The most common autosomal dominant form, ADPKD, is late onset, genetically heterogenous disorder caused by defective genes. Two genes PKD1 (16p), PKD2 (4q) are identified by linkage studies and the presence of an unidentified third locus is suggested [10, 11, 12, 13]. PKD1 localized in terminal region of chromosome16 having highly repetitive GC rich sequence and extremely prone to genetic rearrangement. It consists of 46 exons spanning 54Kb genomic region transcribes 14.1Kb mRNA of 12,909bp long coding sequence which translates 4,303 amino acids long 462kDa glycosylated, an integral membrane protein known as polycystin1 (PC1). Functional PKD1 gene transcribe two isoforms differ in one amino acid length is produced by alternative splicing of mRNA [27]. The first 33 exons of PKD1 have undergone intra-chromosomal duplication throughout human evolution and generate six pseudogenes 13-16 MB proximal to PKD1 gene on same chromosome. Three pseudogenes viz. HG-A (21Kb), HG-B (17Kb), HG-C (8.5Kb) transcribe and share >95% sequence homology to PKD1 [14, 28, 29]. Because of having cryptic stop codons, they are expected to give small proteins that may be functional or nonfunctional [28]. PKD1 gene encodes three major components of protein (i) extracellular N- terminal is coded by exons 1-25, (ii) the middle transmembrane region is encoded by exon 26 and exons 29-45 and (iii) intracellular C-terminal region by exon 45- 46 [29]. The second gene PKD2 contains 15 exons acquires a 68Kb genomic region transcribes 5.3kb mRNA of 2904bp long coding sequence which encodes 968aa long 110kDa integral membrane protein polycystin2 (PC2). PKD2 gene encodes two major segments (i) transmembrane region and (ii) intracellular region having both N and C terminals. Five isoforms are produced by alternative splicing of mRNA [30, 31]. Exon1 is 660bp long and is very GC- rich. Exons 1-2 encodes N- terminal region, exons 2-9 codes for transmembrane domains, exons 11-15 form C-terminal region and exons 3,4 and 5 codes for extracellular loop.PC2 is member of polycystin subfamily belongs to transient receptor potential superfamily has 6 membrane spanning domains and cytoplasmic N and C terminals. 450aa sequence of PC1 is highly similar to PC2 transmembrane region. This transmembrane region in PC2 form transient cation receptor potential channel and for PC1 it is suspected to have same function [32, 33]. Mutational screenings of the candidate genes suggest about 85% cases are linked with PKD1 gene mutations and rest 15% are linked with PKD2 gene and an unidentified gene mutation [34]. To date ADPKD mutation database listed 436 and 115 DNA sequence variants in PKD1 and PKD2 respectively [35]. Several genetic mutations have been found universally spread through- out the candidate genes. It has been found that single nucleotide changes are more in frequency than large deletions, duplications and insertions [36]. PKD1 is highly polymorphic with about 10 neutral variants found in single patient from sequencing [37]. Some other phenomenon like loss of heterozygosity, de novo mutations, mosaicism and hypomorphic allele are reported from familial cases which suggested alteration in the disease phenotype [38, 39]. Mouse knockouts of both alleles pkd1 and pkd2 attribute embryonic lethality suggesting that mutation in both genes gives severity to the disease [40].
Polycystins
Immunohistology for PC1 explore that it is highly expressed in fetal kidneys and lower in adult kidneys. At organ level it is also found in brain, liver, pancreas, heart, intestine and cell lines such as HEK293T [41]. Cellular expression of PC1 is highly confined to epithelial cells and at subcellular level it is localized at plasma membrane. In >20 weeks old fetus and adult kidneys PC1 was mainly localized at loop of Henle and cortical tubules. In adult kidneys it is more localized to cell-cell junction, cilia, apical and basolateral portion of collection duct epithelial cells [41, 42, 43]. PC1 encoded by PKD1 is cardinal member of a novel class of proteins involved in developmental processes [44]. PC1 has evolutionary conserved domains of multiple functions [45]. PC1 regulates calcium ion influx via interaction of coiled coil domains of PC1 and PC2 [46]. Similarly, PC2 expressed in kidney, heart, ovary, testis, vascular smooth muscles and small intestine. This protein is expressed in all segments of nephron except glomeruli. Sub-cellular localization of PC2 is confined to Golgi compartment, endoplasmic reticulum, and plasma membrane. PC2 share structural feature with transient receptor potential (TRP) channel as well as voltage activated calcium and sodium channel. It can form homo and hetero multimer with the help of its C-terminal tail containing two different sites for PC2 and PC1 protein- protein interactions respectively [47]
Cystogenesis
Molecular studies performed in animal models and human kidneys reveals that several proteins are involved in cystogenesis and among all these two ciliary proteins PC1 and PC2 are found prominently. Genetic studies on polycystic kidney disclose that occurrence of renal cyst in many disorders expose multiple molecular signaling pathways regulating different cell processes and maintenance of extracellular environment which get dysregulated in cystogenesis [48]. Non-genetic and simple cystic disorders suggest the involvement of other modifier genes and environmental factors in cystogenesis independent of age and gender of individual [49]. Cyst formation starts from all parts of nephron at teen-age and with the age cyst number and size enlarges. Cysts spread throughout the cortex and medulla leading to disruption of renal parenchyma of kidney and become prominent fluid filled saccular entity at later stage of life. Bud like outgrowth formed due to uncontrolled cell proliferation of epithelium lining of nephron which shed off from parent nephron and form independent sac like structure [50]. Genetic defects containing epithelium cells have abnormal cell secretory nature which leads to expansion of sac. Presence of somatic mutations in epithelial cells of cultured cyst suggested “two hit mechanism” of cystogenesis in which inactivation of both copies of a candidate gene is required for cyst formation. The “first hit” is inherited through germ line mutation, whereas the “second hit” is somatic mutation acquired during life time elicit cyst formation [26, 51]. Intrafamilial phenotypic variation suggests the involvement of some other modifying internal factors such as gene/s, metabolic products in this disease which can be targeted in disease management. Interfamilial pathophysiological variations suggest us searching of other external modifiers such as environment and unidentified gene/s aggravate the cyst formation [52]. Molecular studies performed in mouse model suggested contribution of Pkd1 gene dose like low expression [53], overexpression [54] in cystogenesis. Microsatellite marker-based studies performed on familial cases suggested the involvement of loss of heterozygosity cystogenesis [38]. Experiments on PKD null mice indicate that PC1 play important role in renal tubule maturation [55]. Overlapping of sub-cellular localization of PC1 and PC2 suggest their mutual function in different signaling pathways [48]. Disruption in polycystin resulted in the uncontrolled cellular programs such as cell proliferation, differentiation, apoptosis, cell polarity, adhesion, maturation and extracellular matrix maintenance which are all essential steps of kidney tubulogenesis and morphogenesis [4]. Gene expression studies on mouse model and ADPKD human kidney samples identified differentially expressed genes involved in wnt/beta catenin, receptor tyrosine kinase and G- protein coupled signaling which get dysregulate in cystogenesis [56, 57, 58]. PC1 and PC2 are ciliary proteins function as mechanosensor molecule to correctly sense luminal flow. Defect in the proteins unable to sense luminal flow induce the cystogenesis [59]. Renal tubular epithelium proliferation is controlled process but failure in cilium or basal body/centrosome complex assembly, play important role in mitotic tubules orientation during cell division could trigger uncontrolled cell proliferation [60]. Confirmation study of imbalance between the cell division and apoptosis was confirmed as the key factor of cyst growth in Han: SPRD rat model. It inhibits caspases and revealed reduction in tubular apoptosis and slow disease progression [55]. To be fully functional, PC1 must be able to undergo N terminal cleavage which generates the noncovalently attached N terminal region to remaining transmembrane domain. N terminal cleavage occurs at the G protein coupled receptor proteolytic site (GPS) [62]. Two other independent cleavages at C- terminal tail (CTT) of PC1 release ~35kDa and ~15kDa small soluble fragments which binds to intracellular transcription factors and signaling molecules to gene expression and signal transduction. Increased level of cleaved CTT is observed in ADPKD cystic cells [63]. Ribosome biogenesis, protein synthesis and cell size are regulated by mTOR complex. PC1 is involved in inhibition of mTOR cascade in association of TSC1and TSC2 proteins [64]. PC1 and PC2 form heterodimer through their C terminal cytoplasmic tails [65]. PC1 with copartner PC2 increase the p21, regulator protein of cell cycle and cell growth level through JAK and STAT proteins activation [66, 67]. G-protein activation by PC1 and PC2 regulate apoptosis, cell proliferation, cell differentiation and cell adaptation through a complex network of binding proteins and signaling. PC1 activates G-protein alpha subunit and positively regulates activity of cJNK and AP1 transcription factor [68]. G-protein also activates calcineurin which in turn activate and localize NFAT (nuclear factor of activated T cells). PC1 suspected to have role in wnt canonical and wnt non-canonical mediated signaling which regulates gene expression, differentiation and planar cell polarity [69]. Functional analysis of PKD1 transgenic lines revealed that Ig-PKD domains in PC1 mediate cell-cell adhesion [70]. In nephron, PC1 located to cell-cell adhesion junction and focal adhesion junctions, whereas in cystic epithelial cells most of the protein is intracellular [71]. Polycystins regulates the cell polarity and localization of the membrane proteins like Na+/K+- ATPase pump, CFTR pump, EGF receptors, E-cadherins, matrix metalloproteinase and integrins etc. (4). Luminal localization of channel protein by PC1 promotes sodium and water secretion into cyst resulting expansion of cyst and hence disruption of renal parenchyma and compression of normal nephron [72]. These cysts contain all feature of parent nephron but became a single entity which affects the function of nephron which leads to accumulation of waste products in blood. Affected individuals are eventually placed on dialysis and transplant. Role of Pkd1 gene in cyst growth was studied in Pkd1 deletion mouse model and found that Pkd1 gene deficiency does not initiate sufficient autonomous cell proliferation leading to cyst formation. It suggests that additional stimuli regulating cell homeostasis, differentiation, cell-cell interactions and cell-matrix interactions are also equally mandatory. This could be the result of differences in genetic heterogeneity, allelic heterogeneity and gene environment interactions.
PKD Research in India
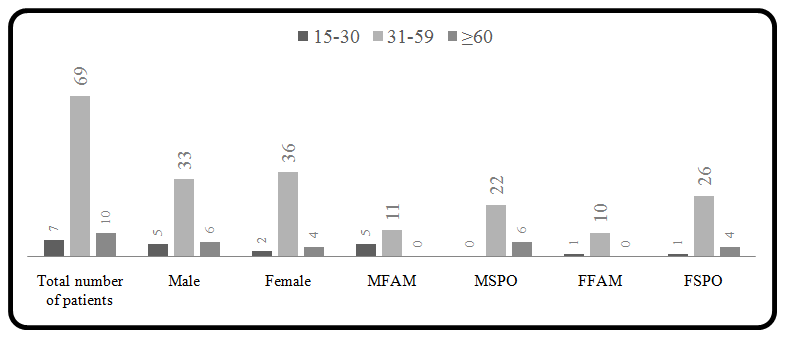
The detailed research work is not available from India for understanding the pathophysiology of PKD. Very recently an India foundation for PKD has been established for the awareness of PKD in Indians and inspire the investigators for basic and applied studies on PKD. A detailed study conducted by us enrolled eighty-four north- Indian individuals and some of the family members were clinically diagnosed with Polycystic Kidney Disease. Based on clinical investigation like family history, symptoms and ultrasonography all recruited patients were confirmed for the disease. Patients with the positive family history were classified as familial case and patients without family history of PKD classified as sporadic cases. Almost equal incidence of PKD in females (48.8%) and males (51.2%) was observed. 30.95% of the total cases had positive family history (familial: FAM) while rest 69.05% had negative family history (sporadic: SPO) (Table 1).
| Sample (blood) | Male | Female | Frequency (%) | Total cases (84) | With family member |
| Sporadic cases (negative family history) | 28 | 30 | 69.04 | 58 | - |
| Familial cases (positive family history) | 15 | 11 | 30.95 | 26 | 22+4(43) |
Table 1: Categorization of patients based on family history and gender.
| 49 PKD patients | Age (years) | Right Kidney-Length (11cm) | Right Kidney–Width (6cm) | Left Kidney-Length (12cm) | Left Kidney-Width (6cm) |
|---|---|---|---|---|---|
| Average ± deviation | 46.6 ± 7.4 | 14.0 ± 2.5 | 6.7 ± 1.5 | 14.5 ± 2.4 | 7.0 ± 1.4 |
Table 2: Size of kidney in PKD cases.
| 49 PKD patients | Age (years) | Right Kidney-Length (11cm) | Right Kidney–Width (6cm) | Left Kidney-Length (12cm) | Left Kidney-Width (6cm) |
|---|---|---|---|---|---|
| Average ± deviation | 46.6 ± 7.4 | 14.0 ± 2.5 | 6.7 ± 1.5 | 14.5 ± 2.4 | 7.0 ± 1.4 |
Table 2: Size of kidney in PKD cases.
hypertension and presence of liver cysts and hepatomegaly were found in 19% and 39% of polycystic patients respectively (table 3).Clinical symptoms and the family history suggested that the patients might have favorable genetic background responsible for disease condition.
| Clinical Symptoms in PKD cases | Percentage |
|---|---|
| Liver cyst and hepatomegaly | 39 |
| Hypertension | 19 |
| Chronic kidney disease | 17 |
| Kidney stone | 12 |
| Type 2 diabetes mellitus | 8 |
| Gall bladder stone | 6 |
| Lymphadenopathy | 5 |
| Urinary tract infection | 5 |
| Hematuria | 5 |
| Prostatic cyst | 5 |
| Ascites | 5 |
| Benign prostate hyperplasia | 4 |
| Hypothyroidism | 2 |
| Liver stone | 2 |
| Cardiac valve defect | 2 |
| Asthma | 2 |
| Pancreatic cyst | 1 |
| Other symptoms | 17 |
Table 3: Frequency of clinical symptoms in Indian PKD cases.
ADPKD is a slow progressive genetic disorder which could be treated by retarding the cyst expansion. Prevalence and susceptibility of disease depends of genotype of population and external environmental factors. There are several case reports published from all over the world to explain the exact scenario of ADPKD so that the genotype can be correlated with phenotype. A better understanding of the epidemiology and etiology of polycystic kidney disease will reveal the possible therapy and management of disease progression.
Acknowledgment
This work was supported by UGC-UPE focus area-II, Banaras Hindu University, INDIA.
References
-
Zerres K, Völpel MC, WeiB H (1984) Cystic kidneys: Genetics, pathologic anatomy, clinical picture, and prenatal diagnosis. Hum Genet 68(2): 104-135.
-
Gardner KD (1988) Cystic kidneys. Kidney Int 33(2): 610-621. autosomaldominant and recessivepolycystic kidney diseases. Nat Clin Pract Nephrol 2(1): 40-55.
-
Torres VE, Harris PC, Pirson Y (2007) Autosomal dominant polycystic kidney disease. Lancet 369(9569): 1287-1301.
-
Breuning MH, Snijdewint FG, Brunner H, Verwest A, Ijdo JW, et al. (1990) Map of 16polymorphicloci on the short arm of chromosome16close to the polycystic kidney disease gene (PKD1). J Med Genet 27(10): 603-613.
-
Coto E, de Castro SS, Aguado S, Alvarez J, Arias M, et al. (1995) DNA microsatellite analysis of families with autosomal dominant polycystic kidney disease types1 and 2: evaluation of clinical heterogeneity between both forms of the disease. J Med Genet 32(6): 442- 445.
-
Iglesias DM, Martín RS, Fraga A, Virginillo M, Kornblihtt AR, et al. (1997) Genetic heterogeneity of autosomal dominant polycystic kidney disease in Argentina. J Med Genet 34(10): 827-830.
-
Jeffery S, Morgan S, Warmington VJ, MacGregor GA, Saggar Malik AK (1995) A family with autosomal dominant polycystic kidney diseaselinked to 4q21-23. J Med Genet 32(6): 493-494.
-
Reeders ST, Breuning MH, Corney G, Jeremiah SJ, Meera Khan P, et al. (1986) Twogenetic markers closely linked to adult polycystic kidney disease on chromosome 16. Br Med J 292(6524): 851-853.
-
de Almeida E, Prata MM, de Almeida S, Lavinha J (1999) Long-term follow-up of a family with autosomal dominant polycystic kidney disease type3. Nephrol Dial Transplant 14(3): 631-634.
-
Ariza M, Alvarez V, Marín R, Aguado S, Coto E, et al. (1997) A family with a milder form of adult dominant polycystic kidney disease not linked to the PKD1 (16p) or PKD2 (4q) genes. J Med Genet 34(7): 587- 589.
-
Daoust MC, Reynolds DM, Bichet DG, Somlo S (1995) Evidence for a third genetic locus for autosomal dominant polycystic kidney disease. Genomics 25(3): 733-736.
-
Paterson AD, Pei Y (1998) Is there a third gene for autosomal dominant polycystic kidney disease? Kidney Int 54(5): 1759-1761.
-
Rossetti S, Strmecki L, Gamble V, Burton S, Sneddon V, et al. (2001) Mutation analysis of the entirePKD1gene: genetic and diagnostic implications. Am J Hum Genet 68(1): 46-63
-
Guyton AC, Hall JE (2006) Urine formation by the kidneys: I glomerular filtration, renal blood flow, and their control. Textbook of Medical Physiology. Philadelphia, Pennsylvania: Elsevier Saunders 11:307-326.
-
Casal JA, Hermida J, Lens XM, Tutor JC (2005) A comparative study of three kidney biomarker tests in autosomal dominant polycystic kidney disease. Kidney Int 68: 948-954.
-
Boucher C, Sandford R (2004) Autosomal-dominant polycystic kidney disease (ADPKD, MIM173900, PKD1 and PKD2genes, protein products known as polycystin-1 and polycystin-2). Eur J Hum Genet 12(5): 347-354.
-
Shamshirsaz AA, Reza Bekheirnia M, Kamgar M, Johnson AM, McFann K, et al. (2005) Autosomal- dominant polycystic kidney disease in infancy and childhood: progression and outcome. Kidney Int 68(5): 2218-2224.
-
D'Cruz S, Singh R, Mohan H, Kaur R, Kopoor V, et al. (2010) Autosomal dominant polycystic kidney disease with diffuse proliferative glomerulonephritis - an unusual association: a case report and review of the literature. J Med Case Rep 4: 125.
-
Saxena S, Hotchandani RK, Bhuyan UN, Agarwal SK, Tiwari SC, et al. (1993) Membranous glomerulonephritis associated with autosomal dominant polycystic kidney disease. Nephron 65(2): 316-317.
-
Kumar S, Nanjappa B, Barapatre YR (2012) Autosomal dominant polycystic kidney disease with situs inversus. Urology 80(2): e23-24.
-
Johnson DK, Panchili S, Kolasseri S, Mavali RT (2014) Polycystic liver disease presenting as pruritus: Annals of Gastroenterology 27(1): 76-78.
-
Wani I, Kirmani O (2013) Traumatic intracystic hemorrhage in polycystic kidney. JCRP 2: 47-48.
-
Shanmuganathan A, Rao UK, Chandrasekharan A, Thanasekaraan LD, Palanisamy CA, et al. (2008) Recurrent pneumothorax associated with polycystic kidney disease. Indian J Chest Dis Allied Sci 50: 233- 235.
-
Radpour R, Rezaee M, Haghighi MM, Ohadi M, Najmabadi H, et al. (2006) Genetic heterogeneity of PKD1 and PKD2 genes in Iran and determination of the genotype/phenotype correlations in several families with autosomal dominant polycystic kidney disease. IBJ 10(1): 1-8.
-
Watnick TJ, Torres VE, Gandolph MA, Qian F, Onuchic LF, et al. (1998) Somatic mutation in individual liver cysts supports a two-hitmodel of cystogenesis in autosomal dominant polycystic kidney disease. Mol Cell 2(2): 247-2451.
-
Wołyniec W, Jankowska MM, Król E, Czarniak P, Rutkowski B (2008) Current diagnostic evaluation of autosomal dominant polycystic kidney disease. Pol Arch Med Wewn 118(12): 767-773.
-
(1995) Polycystic kidney disease: the complete structure of the PKD1 gene and its protein. The International Polycystic Kidney Disease Consortium. Cell 81(2): 289-298.
-
Symmons O, Váradi A, Arányi T (2008) How segmental duplications shape our genome: recent evolution of ABCC6 and PKD1Mendelian disease genes. Mol Biol Evol 25(12): 2601-2613.
-
Thongnoppakhun W, Wilairat P, Vareesangthip K, Yenchitsomanus PT (1999) Long RT-PCR Amplification of the entire coding sequence of the polycystic kidney disease1 (PKD1) gene. Biotechniques 26(1): 126-132.
-
Hayashi T, Mochizuki T, Reynolds DM, Wu G, Cai Y, et al. (1997) Characterization of the exonstructure of the polycystic kidney disease 2gene (PKD2). Genomics 44(1): 131-136.
-
Mochizuki T, Wu G, Hayashi T, Cai Y, Somlo S, et al. (1996) PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 272(5266): 1339-1342.
-
Cantiello HF (2004) Regulation of calcium signaling by polycystin-2. Am J Physiol Renal Physiol 286(6): F1012-F1029.
-
Rossetti S, Harris PC (2007) Genotype-phenotype correlations in autosomal dominant and autosomal recessive polycystic kidney disease. J Am Soc Nephrol 18(5): 1374-1380.
-
Rossetti S, Hopp K, Sikkink RA, Torres VE, Harris PC, et al. (2012) Identification of genemutations in autosomal dominant polycystic kidney disease through targeted resequencing. J Am Soc Nephrol 23(5):915-933.
-
Reed B, McFann K, Kimberling WJ, Pei Y, Gabow PA, et al. (2008) Presence of de novo mutations in autosomal dominant polycystic kidney disease patients without family history. Am J Kidney Dis 52(6): 1042-1050.
-
Bataille S, Berland Y, Fontes M, Burtey S (2011) High Resolution Melt analysis for mutation screening in PKD1and PKD2: BMC Nephrology 12: 57-68.
-
Brasier JL, Henske EP (1997) Loss of the polycystic kidney disease (PKD1) region of chromosome 16p13 in renal cyst cells supports a loss-of-function model for cyst pathogenesis. J Clin Invest 99(2): 194-199.
-
Reiterová J, Štekrová J, Merta M, Kotlas J, Elišáková V, et al. (2013) Autosomal dominant polycystic kidney disease in a family with mosaicism and hypomorphic allele. BMC Nephrol 14: 59-64.
-
Garcia-Gonzalez MA, Outeda P, Zhou Q, Zhou F, Menezes LF, et al. (2010) Pkd1 and Pkd2 are required for normal placental development. PLoS ONE 5(9).
-
Chae SW, Cho EY, Park MS, Lee KB, Kim H, et al. (2006) Polycystin-1 expression in fetal, adult and autosomal dominant polycystic kidney. J Korean Med Sci 21(3): 425-429.
-
Geng L, Segal Y, Peissel B, Deng N, Pei Y, et al. (1996) Identification and localization of polycystin, the PKD1 Gene Product. J Clin Invest 98(12): 2674-2682.
-
Ward CJ, Turley H, Ong ACM Comley M, Biddolph S, et al. (1996) Polycystin, the polycystic kidney disease 1 protein, is expressed by epithelial cells in fetal, adult, and polycystic kidney. Proc Natl Acad Sci 93(4): 1524-1528.
-
van Adelsberg JS (1999) The role of the polycystins in kidney development. Pediatr Nephrol 13(5):454-459.
-
Sandford R, Sgotto B, Aparicio S, Brenner S, Vaudin M, et al. (1997) Comparative analysis of the polycystic kidney disease 1 (PKD1) gene reveals an integral membrane glycoprotein with multiple evolutionary conserved domains. Hum Mol Genet 6(9): 1483-1489.
-
Casuscelli J, Schmidt S, DeGray B, Petri ET, Celić A, et al. (2009) Analysis of the cytoplasmic interaction between polycystin-1 and polycystin-2. Am J Physiol Renal Physiol 297(5): F1310-F1315.
-
Tsiokas L, Kim E, Arnould T, Sukhatme VP, Walz G (1997) Homo- and heterodimericinteractions between the geneproducts of PKD1 and PKD2. Proc Natl Acad Sci 94(13): 6965-6970.
-
Vandorpe DH, Wilhelm S, Jiang L, Ibraghimov- Beskrovnaya O, Marina N, et al. (2002) Cation channel regulation by COOH-terminal cytoplasmic tail of polycystin-1: mutational and functional analysis. Physiol Genomics 8(2): 87-98.
-
Yoder BK, Hou X, Guay-Woodford LM (2002) The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol 13(10): 2508-2516.
-
Fain PR, McFann KK, Taylor MR, Tison M, Johnson AM, et al. (2005) Modifier genes play a significant role in the phenoltypic expression of PKD1. Kidney Int 67(4): 1256-1267.
-
Harris PC (2010) What is the role of somatic mutation in autosomal dominant polycystic kidney disease? J Am Soc Nephrol 21(7): 1073-1076.
-
Pie Y (2005) Nature and nurture on phenotypic variability of autosomal dominant polycystic kidney disease. Kidney Int 67(4): 1630-1631.
-
Lantinga-van Leeuwen IS, Dauwerse JG, Baelde HJ, Ward CJ, Verbeek S, et al. (2004) Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum Mol Genet 13(24): 3069-3077.
-
Thivierge C, Kurbegovic A, Couillard M, Guillaume R, Coté O, et al. (2006) Overexpression of PKD1causespolycystic kidney disease. Mol Cell Biol 26(4): 1538-1548.
-
Boletta A, Germino GG (2003) Role of polycystins in renal tubulogenesis. Trends Cell Biol 13(9): 484-492.
-
Chen WC, Tzeng YS, Li H (2008) Gene expression in early and progression phases of autosomal dominant polycystic kidney disease. BMC Research Notes 1: 131-140.
-
Pandey P, Qin S, Ho J, Zhou J, Kreidberg JA (2011) Systems biology approach to identify transcriptome reprogramming and candidate microRNA targets during the progression of polycystic kidney disease. BMC Syst Biol 5: 56-78.
-
Song X, Di Giovanni V, He N, Wang K, Ingram A, et al. (2009) Systems biology of autosomal dominant polycystic kidney disease (ADPKD): computational identification of gene expression pathways and integrated regulatory networks. Hum Mol Genet 18(13): 2328-2343.
-
Kotsis F, Boehlke C, Kuehn EW (2013) The ciliary flow sensor and polycystic kidney disease. Nephrol Dial Transplant 28(3): 518-526.
-
Simons M, Walz G (2006) Polycystickidneydisease: celldivision without a c(l)ue? Kidney Int. 2006; 70(5): 854-864.
-
Kugita M, Nishii K, Morita M, Yoshihara D, Kowa- Sugiyama H, et al. (2011) Global gene expression profiling in early-stage polycystic kidney disease in the Han:SPRD Cy rat identifies a role for RXR signaling. Am J Physiol Renal Physiol 300(1): F177- F188.
-
Chapin HC, Rajendran V, Caplan MJ (2010) Polycystin- 1 surface localization is stimulated by polycystin-2 and cleavage at the G protein-coupled receptor proteolytic site. Mol Biol Cell 21(24): 4338-4348.
-
Bertuccio CA, Caplan MJ (2013) Polycystin-1 C terminus cleavage and its relation with polycystin-2, two proteins involved in polycystic kidney disease. Medicina 73(2): 155-162.
-
Rosner M, Hanneder M, Siegel N, Valli A, Fuchs C, et al. (2008) The mTOR pathway and its role in human genetic diseases. Mutation Research 659: 284-292.
-
Talbot JJ, Shillingford JM, Vasanth S, Doerr N, Mukherjee S, et al. (2011) Polycystin- 1regulatesSTATactivity by a dual mechanism. Proc Natl Acad Sci 108(19): 7985-7990.
-
Weimbs T, Olsan EE, Talbot JJ, (2013) Regulation of STATs by polycystin-1 and their role in polycystic kidney disease. Jakstat 2(2): e23650-e23659.
-
Parnell SC, Magenheimer BS, Maser RL, Zien CH, Frischauf AM, et al. (2002) Polycystin-1 activation of c-Jun N-terminal kinase and AP-1 is mediated by heterotrimeric G proteins. JBC 277(22): 19566- 19572.
-
Puri S, Magenheimer BS, Maser RL, Ryan EM, Zien CA, et al. (2004) Polycystin-1activates the calcineurin/NFAT (nuclearfactor of activated T-cells) signaling pathway. J Biol Chem 279(53): 55455- 55464.
-
Chapin HC, Caplan MJ (2010) The cellbiology of polycystic kidney disease. J Cell Biol 191(4): 701-710.
-
Streets AJ, Newby LJ, O'Hare MJ, Bukanov NO, Ibraghimov-Beskrovnaya O, et al. (2003) Functional analysis of PKD1 transgenic lines reveals a direct role for polycystin-1 in mediating cell-cell adhesion. J Am Soc Nephrol 14(7): 1804-1815.
-
Wilson PD (2004) Polycystic kidney disease. N Engl J Med 350(2): 151-164.
-
Persu A, Devuyst O (2000) Transepithelial chloride secretion and cystogenesis in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 15(6): 747-750.
- Measuring What Matters: Data Gaps, Certificate of Need Reform, and Pediatric Psychiatric Inpatient Capacity in North Carolina
- Intersecting Epidemics and Climate Vulnerabilities in Conflict- Driven Displacement: Epidemiology, Systemic Challenges, and One Health Gaps in South Sudan
- Advancing Domestic Health Financing for Community Health System Sustainability in South Sudan: The Boma Health Initiative Model (2025–2035)
- Prevalence and Correlates of Post-Exposure Prophylaxis Uptake among Men Who Have Sex with Men in Kisumu County, Kenya
- Medical, Ethical, and Legal Conflicts Surrounding Euthanasia in Argentina. Its Global Implications
- Knowledge and Attitude on Menstrual Hygiene among Adolescent Girls Studying in Secondary Level in Public Schools of Chitwan District, Nepal