Thermoelectrics of MoS2(1-x)N2x Compounds
The electronic and transport properties are calculated for the layered binary transition metal compounds Molybdenum disulphide and dinitride MoS2(1-x)N2x for x=0 and 1 respectively; as well as the ternary compounds formed with ‘x’ taking values between 0 to 1 by using the density functional theory (DFT) and the semi-classical Boltzmann transport theory. All the ternary compounds formed by a systematic substitution of sulphur with nitrogen atoms in MoS2 are interestingly semi metals as seen from their bands structure plots. Moreover, the thermoelectric figure of merit ZT shows a maximum value of around one for the case of the ternary compound MoSN configuration (a) and the molybdenum dinitride (MoN2) at room temperature. The very attractive ZT values predicted below 400K is a good indicator of the thermoelectric capability of these compounds at practical temperatures.
Introduction
Only, a third of all energy input is utilized in most industrial and other processes and the rest is emitted to the environment as ‘waste heat’ energy. In addition, 75% of this waste energy is in the 100-300°C range. Thermoelectric (TE) generators that can harvest this heat energy are a very attractive solution for sustainable energy. This motivates the quest for high performance TE materials that can convert waste heat energy to electricity with minimum energy loss and cost. The key advantages of TE generators are long life, lack of moving parts, low breakdowns and surviving harsh environments.
The past decade has shown the high potential of transition metal dichalcogenides (TMD) for electronic, sensing, photonic and thermoelectric device applications [1, 2, 3]. The application of these materials for thermoelectric energy generation is due to the highly crystalline layered structure with the possibility of easy low dimensional fabrication, the absence of dangling bonds and favorable electronic properties which can lead to significantly large figures-of-merit. The recent comprehensive review by Zhang, et al. [2] shows the excellent suitability of TMDs for thermal management and TE applications. MoS2 as a prototype TMD material has attracted a lot of attention and has been studied and characterized extensively for structural, electronic, magnetic, optical [3, 4, 5] and transport properties both in bulk and in the 2D limit [6, 7]. MoS2 is a prime candidate for TE applications and the TE properties of MoS2 in bulk and in different nano-structural forms like monolayers (ML), bilayers (BL), nanowires etc have been investigated both theoretically and experimentally [7, 8, 9]. The sole purpose in all these interesting works on TE is increasing the heat to electric energy conversion efficiency which is measured by the value of ZT defined as
2 / ZT S T k σ = (1)
S is the Seebeck coefficient, σ is the electrical conductivity and T is the temperature in Kelvin. The total thermal conductivity κ consists of an electronic part κe and a lattice part κ_l. There are many ways to enhance _ZT by optimizing the power factor (PF) S2σ and κ to their maximum and minimum values respectively.
Different strategies have been adopted to achieve super high ZT values like alloying and doping with different atoms [10, 11], changing carrier concentrations [12], nano- structuring [13], and lowering the dimensionality of the systems [7, 8, 9, 10, 14] as seen in recent literature. High TE performance is observed when going down from the bulk to MLs and other nanostructures [6, 7, 8, 9] due to quantum confinement. Nano-structuring causes enhanced phonon scattering and achieves high ZT via reduction in the lattice thermal conductivity. Alloying is a very effective way of changing the chemical and electronic environment so as to lower the thermal conductivity or obtaining materials with high ZT values. Poudel, et al. [13] show that a peak ZT value of 1.4 at 100°C can be achieved in p-type nanocrystalline bismuth antimony telluride bulk alloy. Bulk materials with 2D structures have a tendency to show low thermal conductivity and high Seebeck coefficient due to their strong anisotropic features and intrinsically short mean free paths [14].
Recent literature [4, 5, 6] on MoS2, confirm that substitution with suitable elements is a very effective strategy to change the charge distribution, fermi levels and the band structure dependent properties. There are many ways of doping and substitution doping is one of the conventional methods that has been used successfully both theoretically and experimentally. For instance, p-type doping has been achieved by substituting Mo atoms with Nb in MoS2 [15].
In this work, we investigate the effect of nitrogen substitution in MoS2 on the TE properties. Substitution of chalcogen sulfur with nitrogen in MoS2 is viable and stable due to the fact that nitrogen and sulphur atoms have comparable sizes. Doping is stable when secured by covalent bonding, as in the present case. The proof of this doping mechanism is the recent experimental finding of Azcatl, et al. [16] of substitutional N doping with covalent bonding in MoS2 upon remote N2 plasma treatment. This method of doping is further confirmed by the experimental study of Yang, et al. [17] who show that dominant N-doping mechanism is a one- to-one substitution of sulfur atoms in MoS2. In their work they have used a high temperature facile treatment to dope MoS2 with nitrogen reaching a high atomic concentration of 41% to activate MoS2 for hydrogen evolution reaction applications.
These recent experimental works provide a strong motivation to study theoretically the effect of nitrogen substitution on the electronic and transport properties of MoS2. The presence of nitrogen is expected to change the band structure and indeed, the high TE performance results obtained are very encouraging. This is, to our knowledge, the first study on the effect of nitrogen in MoS2 for boosting the TE properties. Simulations and predictive quantitative measurements are very important to establish key design and operating features for future thermoelectric devices.
Calculation Method
DFT calculations are performed for the various structures of MoS2(1-x)N2x using the ABINIT software program [18]. LDA Trouller Martins pseudopotentials with core corrections [19] are used for Mo and S. Similar to previous works [20, 21] all structural calculations are performed with a convergence criteria of less than 1x10-6 Ha for the Self Consistent Field (SCF) iterations and a threshold of less than 1 mRy/a.u. for optimization of the geometries. The optimized values of k-point grid and cutoff energy for the plane wave basis set, 8x8x8 and 45Ha respectively, are used for the lattice parameter and relaxation calculations.
The relaxed structures with a sufficiently large number of bands and very dense k-points grids are used to obtain the thermoelectric properties with the BoltzTraP code [22]. This code solves the Boltzmann transport equation under the constant relaxation time (τ) approximation (CRTA). BoltzTrap performs Fourier expansion of the band energies to obtain the transport coefficients and takes into account the symmetry of the system. To investigate the dependencies of the transport properties with temperature, the calculations are done for a temperature range 100-1000K and at an optimal carrier concentration of 1021cm-3.
Under CRTA, the electrical/thermal conductivities and also the power factor are dependent on τ and can be obtained only in terms of τ. Their actual values can be obtained from the experimental values of τ for the same or similar system. Owing to the great uncertainties involved in the determination of the τ value, and the lack of experimental values of τ for the alloys under consideration, we will report our findings for σ and κe in terms of τ. However, S and ZT are independent of τ and their values give us a very good indication of the TE performance of the system.
Results and Discussion
Structural and Electronic
The 2H-MoS2 unit cell has hexagonal symmetry and consists of two formula units of MoS2 or six atoms; two Mo and four S atoms, arranged in two stacks of 3 atomic layers consisting of a Mo atomic plane sandwiched between two S atomic planes. The atoms are bonded covalently in plane and the stacks are held together by weak Van der Waals force. The compounds under investigation are formed by one, two and four N replacing S atoms in MoS2 unit cell as represented by the formula MoS2(1-x)N2x (x=0, 0.25, 0.5 and 1). Figure 1
shows some representative structures for x=0, pristine MoS2, the ternary alloys for x=0.5 (two configurations), denoted by MoSN(a) and (b) and MoN2 (x=1) in hexagonal symmetry. The Mo atoms are in cobalt blue, the S atoms in yellow and the N atoms in blue. In configuration MoSN(a) the nitrogen and sulphur atoms are in alternate positions in the unit cell, whereas, configuration (b) has nitrogen terminated ends.
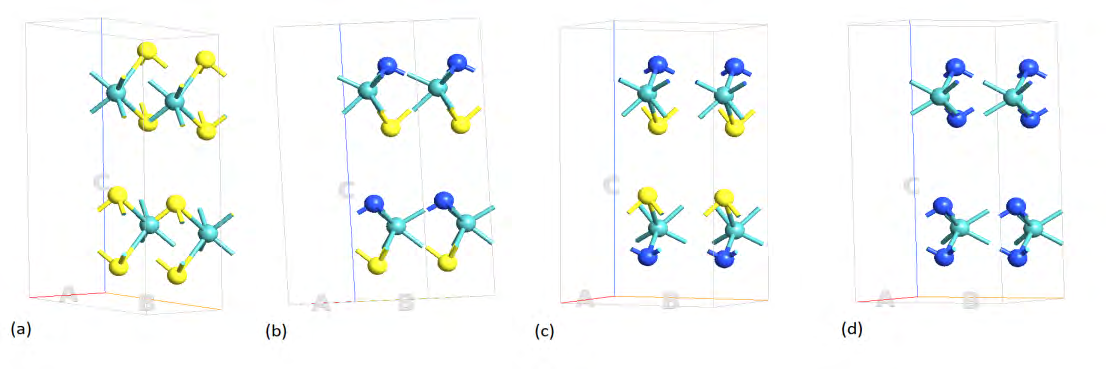
Figure 1: The pure MoS2 and alloyed structures used in the simulation for electronic and transport properties (a) pure MoS2 which consists of a 3-dimensional array of atoms of layers S-Mo-S in a hexagonal lattice (b) 50% N atom substitution for S labeled as MoSN(a) with alternating S and N positions and (c) MoSN(b) with N terminated ends (d) complete substitution of S atoms with N.
It is interesting to note that molybdenum dinitride has been successfully synthesized in the bulk form by Wang, et al. [23] for hydrogenation applications. They found that MoN2 adopts a rhombohedral R3m structure, isotypic with MoS2 and has three times the catalytic activity of MoS2. Moreover, MoN2 monolayer has been studied theoretically by Zhang, et al. [24] as high capacity electrode material for metal ion batteries recently. The 1H configuration was found to be the most stable among the structures considered in their study.
The thermodynamic stability is an important factor the existence and synthesis of compounds. We have evaluated the energies of formation (Efor) for the pure and nitrogen substituted hypothetical MoS2 compounds to give an indication of their stability. The energy of formation is the difference between the ground state total energies of the compound and the elemental constituents. All the compounds are stable as shown by negative values of Efor obtained, which are reported in Table 1.
Table 1 also shows the lattice constants, bond lengths and angles of the relaxed structures. In all the discussion that follows the structural and TE results of the pure MoS2 is taken from our previously published paper [8]. The lattice parameters for pure MoS2 is in excellent agreement with the experimental values [25] aexp =3.16Å and cexp = 12.29Å within 2.3 and 0.6% respectively. The lattice constant values of MoS2 are also in good agreement within 1-3% of other theoretical works [26, 27]. We notice from the Table that is a change in the lattice parameter values and a reduction is observed in the ‘a’ and ‘c’ values with increasing nitrogen content. The ternary compound MoSN(a) shows smaller ‘a’ and ‘c’ values then the nitrogen terminated MoSN(b) one.
| System | Lattice parameters (Å) | Bond length(Å) | Angles (°) | E for eV/atom | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| a | c | z | Mo-S | Mo-N | S-Mo-S | S-Mo-N | N-Mo-S | N-Mo-N | ||
| MoS 2 | 3.192 | 12.360 | 0.120 | 2.801 | _ | 82.020 82.020 | _ | _ | _ | -1.90 |
| MoS N 1.5 0.5 | 3.155 | 12.215 | 0.124 | 2.801 | 2.474 | 83.415 | 72.551 | -2.07 | ||
| MoSN(b) | 3.140 | 12.156 | 0.118 | 2.801 | 2.474 | _ | 72.851 | 72.851 | -2.24 | |
| MoSN(a) | 3.127 | 12.105 | 0.136 | 2.801 | 2.474 | _ | 73.497 73.492 | _ | _ | -2.24 |
| MoN 2 | 3.094 | 11.976 | 0.159 | 2.801 | 2.474 | _ | _ | _ | 65.279 65.279 | -2.50 |
Table1: The structural parameters for the different compounds.
This reduction in lattice constants is due to the fact that the introduction of nitrogen changes the chemical environment of the pure MoS2. The complete relaxation of all the atoms to relieve the compressive strain in the system results in a change of the equilibrium minimum energy geometry as reflected in Table1. We notice from the Table that Mo-N bond lengths are shorter than Mo-S bonds and also the angles with nitrogen atoms are smaller. Hence, there is a systematic reduction in the lattice constants values of the structures with increasing nitrogen content. The compressive strain caused by the presence of nitrogen atoms and in agreement with the experimental work of Azcatl, et al. [16]. They doped MoS2 with nitrogen through a remote N2 plasma surface treatment and found the doping mechanism to be substitution of the chalcogen sulfur by nitrogen. Their results give evidence of the covalent nature of the bonding and the introduction of compressive strain in MoS2 by nitrogen substitution. Compressive strain could be the reason for the enhanced TE performance of the N substituted ternary alloys as compared to pure MoS2 in the present work. This conjecture is demonstrated by the DFT calculations of Dimple et al and Battachariya, et al. [28, 29]. In these works, we see how compressive strain plays an important role in modifying the bandgap and improving the TE properties.
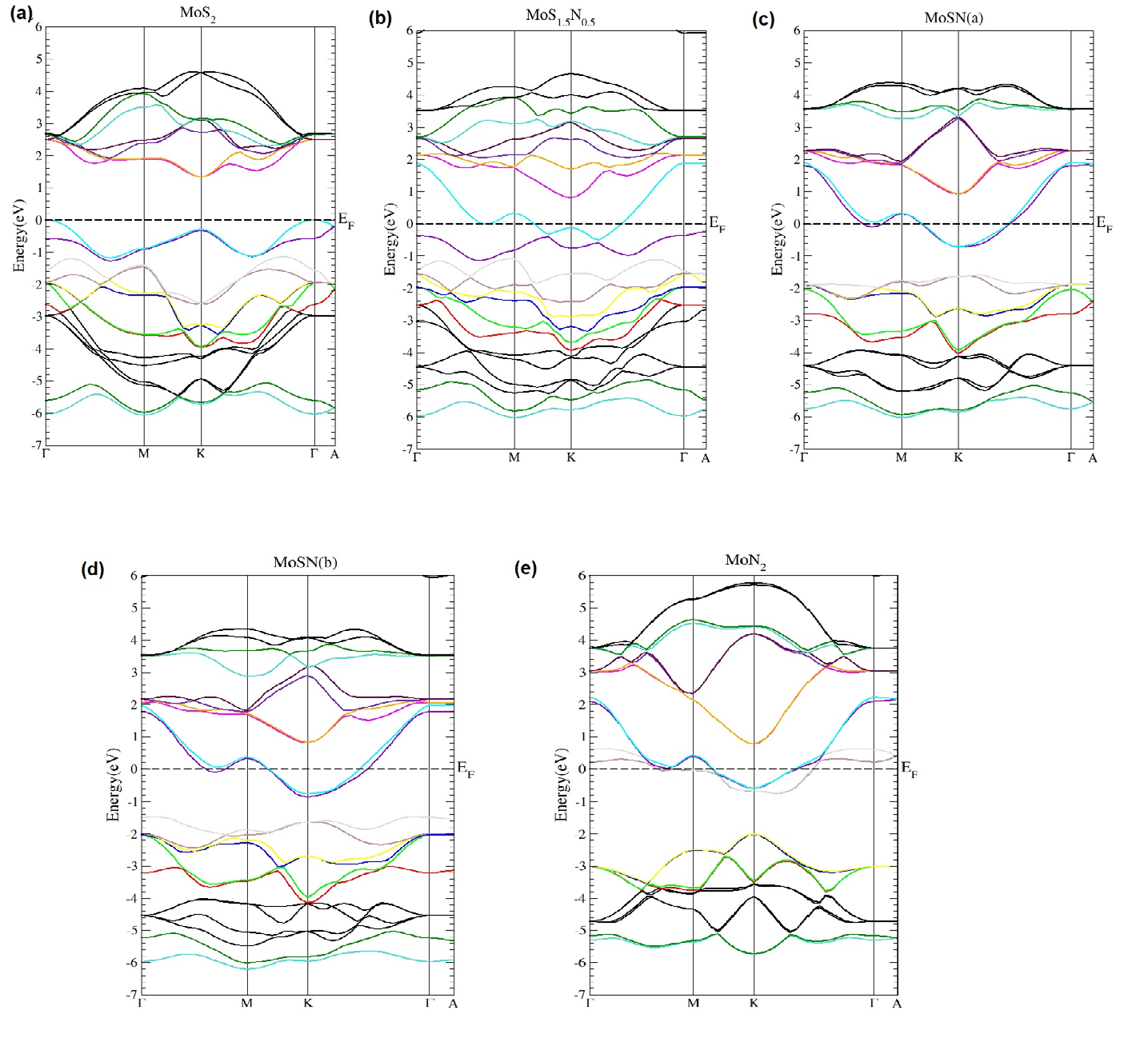
There is also change in the electronic distribution which is also reflected in the band structures for pure MoS2 and the alloyed compounds. The band structures for the relaxed structures of MoS2 and the alloys are depicted in Figure 2 and illustrate the change in behavior from semiconducting to semi metallic with increasing nitrogen concentration. We notice from the band structure for all the compounds other than MoS2, an overlap between the bottom of the conduction band and the top of the valence band, which increases with nitrogen amount. This semi metal feature implies that there is a range of energies for which electrons and holes co-exist. The Mo 4d, S 3p and N 2p atomic orbitals play a decisive role in the band structure properties of pure and nitrogen alloyed compounds of MoS2 and these are the dominant orbitals near the Fermi level. The hybridization of the Mo-d with S-p and N-p orbitals results in a change in the band structure with some conduction bands being pushed down into the valence band and causing the band gap to close in the alloy compounds which exhibit a rich semi-metallic behavior. The pristine MoS2 band structure shows the semiconducting nature and has an indirect Γ− K bandgap of 1.3eV which is in very good agreement with other theoretical and experimental works.
Thermoelectric
The optimized structures of the pristine MoS2 and the nitrogen substituted compounds are used to calculate the electronic band structures at sufficiently dense grids and with a large number of bands and to extract the thermoelectric properties using the BoltzTrap code. Figure 3 shows the variation of the Seebeck coefficient S with the chemical potential at room temperature, 300K.
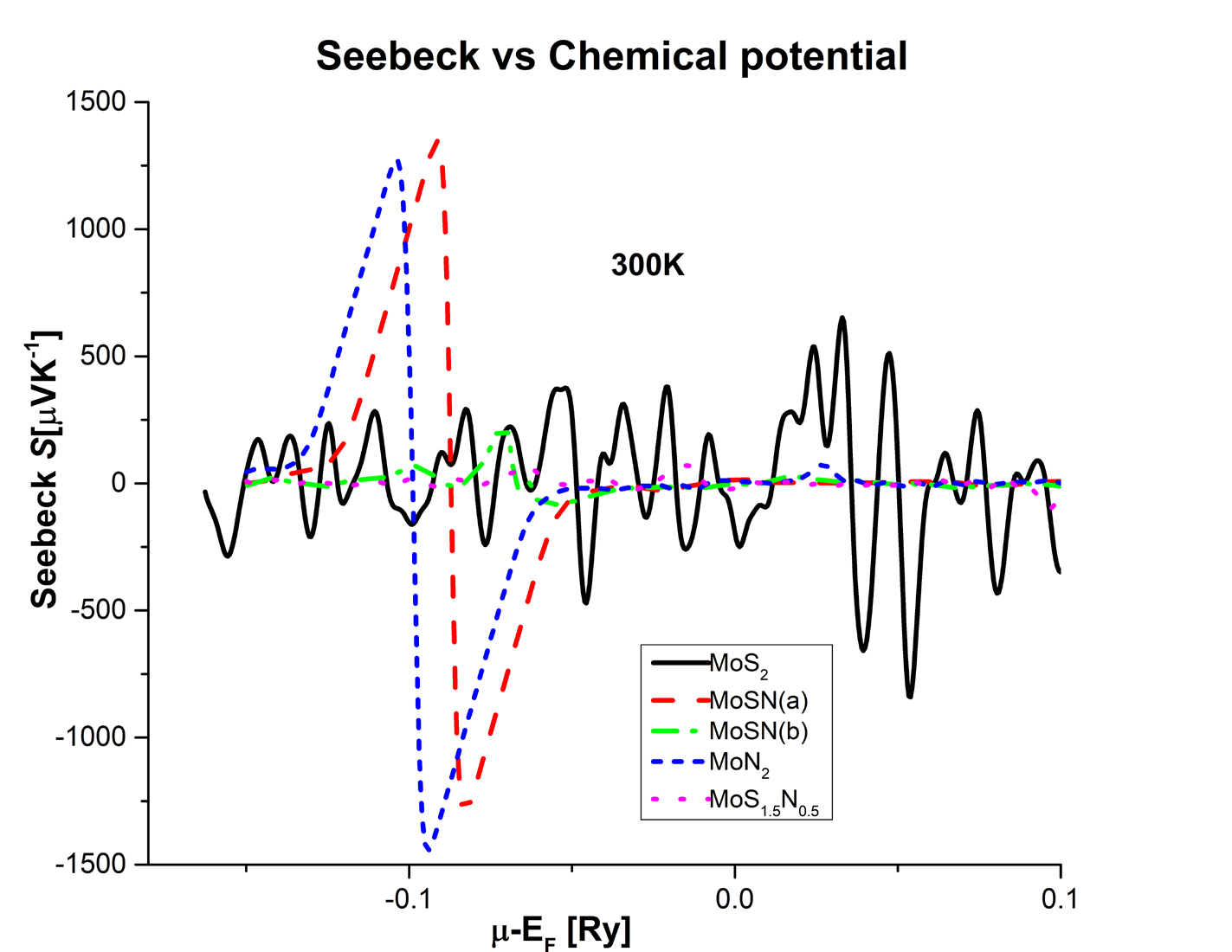
The figure shows both positive and negative large Seebeck values for MoN2 and the MoSN(a) alloy compounds that are around 1500 µV/K as indicated in Table 2. This indicates the high potential of these structures to act as both p and n-type thermoelectrics. Electronic, optoelectronic, and spintronic devices require both n- and p-type materials to form junctions and support bipolar carrier conduction. Usually, only one type of doping is stable for a particular material and the possibility of having bipolar doping is very advantageous in micro and nanoelectronics.
Surprisingly the configuration MoSN(b) has very small Seebeck value, smaller than pure MoS2. The compound MoS1.5N0.5 has the least S values, even more smaller values than MoSN(b) as seen from the graph. This could be due to the lack of symmetry of in this structure which has an odd number of nitrogen atoms. To get a quantitative comparison, the maximum optimal values of thermoelectric properties at room temperature for the different alloys have been tabulated in Table 2.
| Property Structure | S [µV/K] | σ / τ *1019 [Ώms]-1 | κ /τ *1014 e [W/mKs] | Electronic ZT |
|---|---|---|---|---|
| MoS 2 | -840.2 | 2494.1 | 1659.5 | 0.176 |
| MoS N 1.5 0.5 | 101.8 | 23.32 | 18.29 | 0.385 |
| MoSN(a) | 1346.78 | 27.19 | 19.54 | 0.981 |
| MoSN(b) | 241.07 | 19.56 | 13.67 | 0.817 |
| MoN 2 | -1471.02 | 48.97 | 23.96 | 0.968 |
Table 2: The TE maximum values at T=300K for the different properties of the binary and ternary compounds.
We see from the table that the fully N substituted alloy MoN2 has the highest S and second highest ZT value. The S and ZT values of pure MoS2 ~840 µV/K and 0.18 respectively, are well in agreement with other experimental and theoretical works [26, 27]. The nitrogen terminated alloy MoSN(b) has a S value one third that of pure MoS2. But, it also has electrical and thermal conductivities that are two orders of magnitudes smaller than that of MoS2 leading to the appreciable ZT value. All the alloys except MoS1.5N0.5 show a four-fivefold increase in ZT as compared to pure MoS2. The high ZT values are the result of the low electronic thermal conductivities κe/τ of the alloys which drops from ~1700 *1014 W/mKs in MoS2 to a few tens in the alloy compounds. The remarkable decrease in thermal conductivity can be attributed to alloy scattering and changes in band structure due to the presence of nitrogen. On the other hand MoS1.5N0.5 shows a very unfavorable ZT value on account of the very low S values for this compound.
In order to understand better the TE performance, the variations of the electronic thermoelectric properties with temperature are plotted in Figures 4 and 5. The right and left panels show the plots for MoS2 and the nitrogen substituted structures respectively. From Figure 4, we see that the electrical conductivities for all the structures are very close in values and constant over the entire range of temperature.
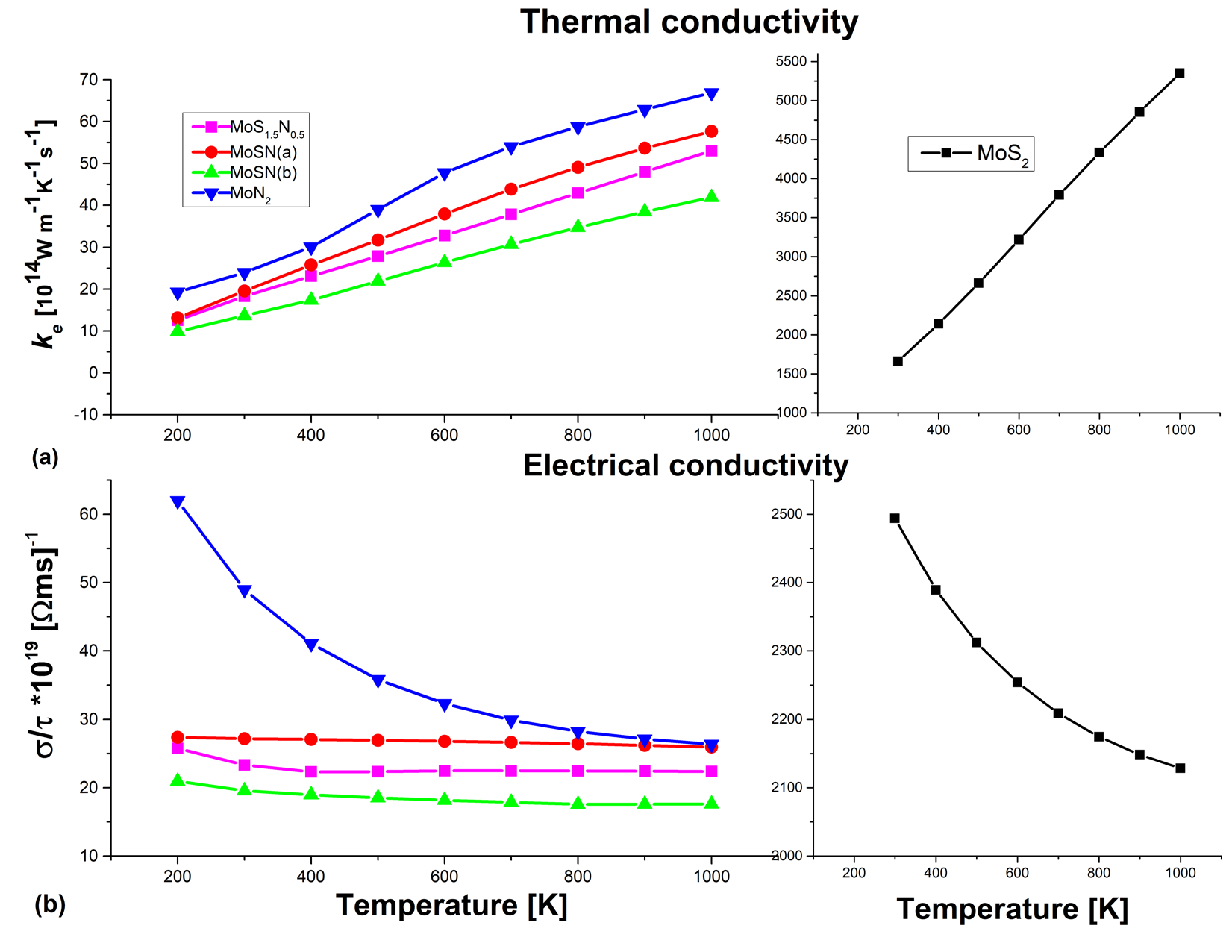
Figure 4 shows the electrical and electronic thermal conductivities as a function of Temperature.
Although, the pure MoS2 also shows an almost constant behavior, the values are ~ two orders of magnitude higher than those of the alloys. We have the same scenario of a flat slope for the κe values of the alloys but pure MoS2 shows a sharp increase in κe with temperature values and is higher by two orders of magnitude. It is this very high thermal conductivity value, coupled with the very low S value that leads to the poor ZT values of bulk MoS2. The maximum values of S and ZT at different temperatures for the different structures are depicted in Figure 5.
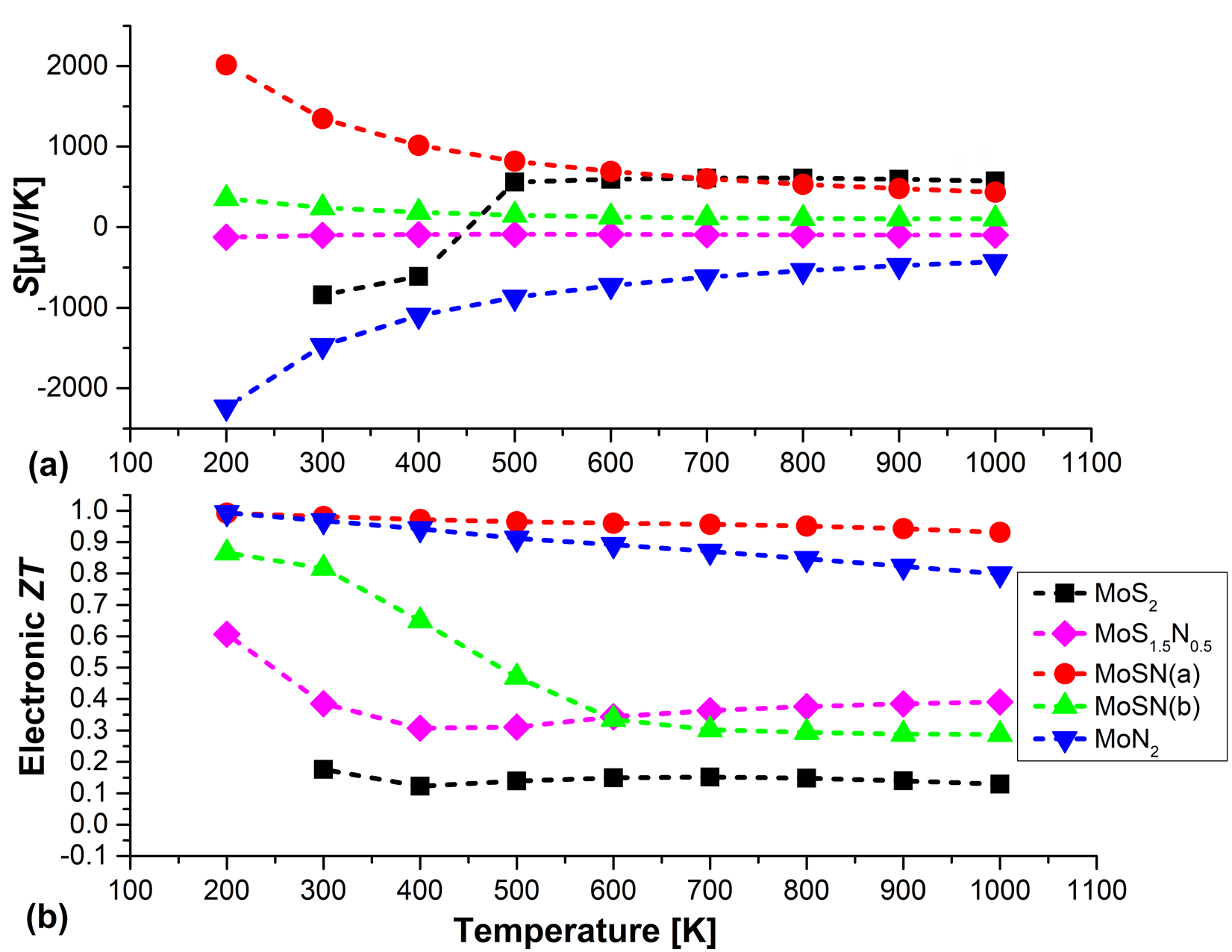
The variation of the maximum values of S with temperature shows that pure MoS2 below 500K has dominant negative S values indicating an n-type TE, whereas above 500K it has positive values and p-type behavior is dominant. With regards to the ternary compounds (x=0.5), there is consistent behavior and both configurations MoNS(a) and (b) and they show dominant positive values with temperature indicating p-type of thermoelectric nature. But, unlike MoSN(a) the nitrogen terminating MoSN(b) has an almost constant and low S values over the entire range of temperature and consequently lower ZT values. In comparison, MoN2 shows dominant n-type behavior with large dominant negative S values at 100k which decreases in magnitude to reach constant values after 500K.
A look at the ZT plot shows that the best thermoelectric performers are MoN2 and MoSN(a) compounds. All three compounds MoN2, MoSN(a) and (b) are good low temperature ≤ 400K thermoelectrics with ZT ranging from 1-0.8. The ZT value of MoS1.5N0.5 although around twice that of MoS2 is the worst TE performer in our study. The general trend is with increasing temperature the ZT values fall, but for the case of the nitrogen terminated MoSN(b) alloy the fall in ZT is rather sharp after 300K and its value is not much enhanced from that of MoS2 above 600K.
Conclusions
Our TE results for nitrogen substitution in MoS2 show the best performance in the low temperature region 200-400K. Maximum ZT is achieved by the MoSN(a) alloy with a value of 0.98, followed closely by the MoN2 alloy with a ZT value of 0.97. However, they show opposite charge carrier behaviors, with MoSN(a) as a dominant p-type and MoN2 as n-type. This is a very useful finding as both p- and n-type TMD materials are highly sought for electrical, optoelectronic, and spintronic applications. They can also be used as the two legs of a single thermo generator that is connected in series electrically and in parallel thermally.
The high S values obtained for the MoN2 and MoSN(a) and (b) alloys are indicative of the high thermopower available for harnessing waste heat energy at low temperatures <400k. This is highly desirable, as in most industrial processes 75%
of the waste heat energy is generated at temperatures <300K.
Acknowledgements
This work was carried out at the theoretical physics laboratory of the Physics Department at the University of Jordan. We are grateful to the deanship of academic research at the University of Jordan for the computational facility and time.
References
-
Wang YH, Huang KJ, Wu X (2017) Recent advances in transition-metal dichalcogenides based electrochemical biosensors: A review. Biosens Bioelectron 97: 305-316.
-
Zhang G, Zhang YW (2017) Thermoelectric properties of two-dimensional transition metal dichalcogenides. J Mater Chem C 5: 7684-7698.
-
Vikraman D, Akbar K, Hussain S, Yoo G, Jang JY, et al. (2017) Direct synthesis of thickness-tunable MoS2 quantum dot thin layers: Optical, structural and electrical properties and their application to hydrogen evolution. Nano Energy 35: 101-114.
-
Ramanathan AA (2018) Defect Functionalization of MoS2 nanostructures as toxic gas sensors: A review IOP Conf Ser Mater Sci Eng 305: 012001.
-
Wang Y, Li S, Yi J (2016) Electronic and magnetic properties of Co doped MoS2 monolayer. Scientific Reports 6: 24153.
-
Kong S, Wu T, Yuan M, Huang Z, Meng QL, et al. (2017) Dramatically enhanced thermoelectric performance of MoS2 by introducing MoO2 nanoinclusions. J Mater Chem A 5: 2004-2011.
-
Jin Z, Liao Q, Fang H, Liu Z, Liu W, et al. (2015) A Revisit to High Thermoelectric Performance of single layer MoS2. Sci Rep 5: 18342.
-
Ramanathan AA, Khalifeh JM (2018) Enhanced thermoelectric properties of suspended mono and bilayer of MoS2 from first principles. IEEE Transactions on nanotechnology 17(5): 974-978.
-
Arab A, Davydov AV, Papaconstantopoulos DA, Li Q (2016) Monolayer MoS2 Nanoribbons as a Promising Material for Both _n_-type and _p_-type Legs in Thermoelectric Generators, Journal of Elec Materi 45: 5253-5263.
-
Ramanathan AA, Khalifeh JM (2017) The electronic and thermoelectric properties of Si1-xVx alloy from first principles. Applied Microscopy 47(3): 105-109.
-
Onofrio N, Guzman DM, Strachan A (2017) Novel doping alternatives for transition metal dichalcogenides from high-throughput DFT calculations. Journal of Applied Physics 122: 185102.
-
Yan Y, Zhang G, Wang C, Peng C, Zhang P, et al. (2016) Optimizing the Dopant and Carrier Concentration of Ca5Al2Sb6 for High Thermoelectric Efficiency. Scientific Reports 6: 29550.
-
Poudel B, Hao Q, Ma Y, Lan Y, Minnich A, et al. (2008) High-Thermoelectric Performance of Nanostructured Bismuth Antimony Telluride Bulk Alloys. Science 320(5876): 634-638.
-
Zhou Y, Zhao LD (2017) Promising Thermoelectric Bulk Materials with 2D Structures. Adv Mater 29(45): 1702676.
-
Suh J, Park TE, Lin DY, Fu D, Park J, et al. (2014) Doping against the Native Propensity of MoS2: Degenerate Hole Doping by Cation Substitution. Nano letters 14(12): 6976-6982.
-
Azcatl A, Qin X, Prakash A, Zhang C, Cheng L, et al. (2016) Covalent Nitrogen Doping and Compressive Strain in MoS2 by Remote N2 Plasma Exposure. Nano Lett 16(9): 5437-5443.
-
Yang Q, Wang Z, Dong L, Zhao W, Jin Y, et al. (2019) Activating MoS2 with Super-High Nitrogen-Doping Concentration as Efficient Catalyst for Hydrogen Evolution Reaction. J Phys Chem C 123(17): 10917- 10925.
-
Gonze X, Jollet F, Araujoa FA, Adams D, Amadon B, et al. (2016) Recent developments in the ABINIT software package. Computer Physics Communications 205: 106- 131.
-
Perdew JP, Wang Y (1992) Accurate and simple analytic representation of the electron-gas correlation energy. Phys Rev B 45(23): 13244.
-
Ramanathan AA, Khalifeh JM (2017) Substrate matters: Magnetic tuning of the Fe monolayer. JMMM 426: 450- 453.
-
Ramanathan AA, Khalifeh JM, Hamad BA (2008) Evidence of surface magnetism in the V/Nb(0 0 1) system: A total energy pseudopotential calculation. Surf Sci 602(12): 607-613.
-
Madsen GKH, Singh DJ (2006) BoltzTraP. A code for calculating band-structure dependent quantities. Computer Physics Communications 175(1): 67-71.
-
Wang SM, Ge H, Sun SL, Zhang JZ, Liu FM, et al. (2015) A New Molybdenum Nitride Catalyst with Rhombohedral MoS2 Structure for Hydrogenation Applications. J Am Chem Soc 137(14): 4815-4822.
-
Zhang X, Yao Y, Yu Z, Wang SS, Guan S, et al. (2016) Theoretical prediction of MoN2 monolayer as a high capacity electrode material for metal ion batteries. J Mater Chem A 4(39): 15224-15231.
-
Coehoorn R, Haas C, Dijkstra J, Flipse CJF, de Groot RA, et al. (1987) Electronic structure of MoSe2, MoS2, and WSe2. I. Band-structure calculations and photoelectron spectroscopy. Phys Rev B 35(12): 6195-6202.
-
Kadantsev S, Hawrylak P (2012) Electronic structure of a single MoS2 monolayer. Solid State Communications 152(10): 909-913.
-
Gandi AN, Schwingenschlogl U (2016) Thermal conductivity of bulk and monolayer MoS2. EPL 113: 36002.
-
Dimple, Jena N, Sarkar AD (2017) Compressive strain induced enhancement in thermoelectric-power-factor in monolayer MoS2 nanosheet. Journal of Physics: Condensed Matter 29: 225501.
-
Bhattacharyya S, Pandey T, Singh AK (2014) Effect of strain on electronic and thermoelectric properties of few layers to bulk MoS2. Nanotechnology 25(46): 465701.
- Sense, Gravity, Parity & Chirality in Mathematical Physics
- Quantum Lattice Simulations PHYSICS: Microcircuit Particle Formation and Observable Macroscopic Irreversible Time - A Discrete Lagrangian with Cellular Automata Framework
- Quantum Biology from Biomacromolecule to Cell, and Central Dogma Described by Quantum Theory
- Focus, Agility, Speed and Technology (FAST) for Sustainability and Growth
- Square Root Metric Geometry and Pati-Salam Model in Curved Space-Time
- A Simple System Demonstrating the Mpemba Effect in Classical Mechanics