Assessment of Awareness, Prevalence and Treatment Trends of Thalassemia in NGO Sector, Sialkot, Pakistan
Thalassemias are the heterogeneous group of genetic disorders affecting hemoglobin production. Purpose: Objective of this study is to access the awareness level among the parents of thalassemia patients, prevalence and treatment trends of thalassemia in NGO sector, Sialkot, Pakistan. Method: Study design was chosen to be cross-sectional prospective. Sample size for this study was taken as 70 patients. Study was carried out in Sundas Foundation in Sialkot, Pakistan. Well-structured data collection form or questionnaire was prepared, analyzed and then filled by interview. Result: Parents of patients were aware of question of iron overload (n=62; %=88.5). Highest prevalence was seen in males (n=42; %=60) rather than females (n=28; %=40). Majority of patients were suffering from thalassemia major (n=60; %=85.7) and few were suffering from thalassemia intermedia (n=10; %=14.3). Hepatitis and iron overload are the two most common complications of thalassemia. Majority of patients (n=69; %=98.5) had iron overload as a complication of blood transfusion, rather than hepatitis (n=43; %=61). Most commonly prescribed iron chelator was deferasirox (n=59; %=84). For therapeutic management of thalassemia, majority of patients were receiving folic acid (n=70; %=100), vitamin-D (n=65; %=92.8), calcium (n=70; % =100), acetaminophen (n= 62; %=88.5) and hydroxyurea (n=12; %=17). From surgical procedures, splenectomy (n=10; %=14) and stem cell transplant (n=1; %=1.4) were used. Conclusion: Highest prevalence was seen in males rather than females.
Introduction
The thalassemias are the set of hereditary hematologic disorders caused by the faults in the generation of one or more of the hemoglobin chains [1]. Thalassemias results from either the reduced synthesis or the complete absence of one or more of the globin chains that constitutes the hemoglobin (Hb) tetramers [2]. Thalassemias are among the most common inherited disorders occurring world- wide, with at least 60,000 individuals born affected every year [3]. Consanguineous marriages are very common in our traditions due to cultural reasons and the family system.
This results in increase in the disease burden with every passing day. Due to our cultural norms, people hesitate to opt for the premarital thalassemia screening even being aware of the nature of its inheritance, having thalassemia in the first cousins and regardless of the current legislations in Pakistan, necessitate pre-marital thalassemia screening for every couple [4]. Carriers of thalassemia mutations are asymptomatic. However, when both parents are carriers, for every pregnancy, there is a 25% chance of having a child with thalassemia, a 50% chance of having a child with thalassemia trait, and a 25% chance of having a normal child. When one parent is carrier, risk of child with thalassemia is 0%, and
50% chances of having a child with thalassemia trait. When one parent is patient and another is carrier, risk of child with thalassemia is 50%, and 50% chances of having a thalassemia carrier. When both parents have thalassemia minor, there is 50% chance of having a child with thalassemia major, 25% chances of having a child with thalassemia minor, and 25% chances of having a normal child [5]. Pakistan has a large population of over 150 million people with a normal carrier frequency of approximately 5.6% of β-thalassemia. Punjab is the largest province in the country with a population of over 50%. The incidence of β - thalassemia is alarming as the local capacity is very high (>81%) and the learning rate is low in South Punjab [6].
Treatment of thalassemia major includes regular RBC transfusions, iron chelation and management of secondary complications of iron excess. In some conditions, spleen removal may be obligatory. Bone marrow transplantation leftovers the only ultimate cure at this time available. Persons with thalassemia intermedia may need splenectomy, folic acid supplementation, treatment of extra medullary erythropoietic masses and leg ulcers, anticipation and therapy of thromboembolic events [7]. Good nutrition with sufficient vitamins and trace elements ingestion, all along with calcium and vitamin D supplementation, can increase bone density and avoid bone loss [8]. Desferroxamine has been found to be widely used in the treatment of thalassemia, restores toxins and prolongs life when used properly. However, the drug is a problem because it can be irritating when injected locally. After years of monitoring and monitoring patients, it is now clear that desferrioxamine can be toxic if given in high doses. There can be serious neurological consequences. Deferiprone, or L1, is the primary oral chelator now used in clinical trials worldwide. The purpose of these drugs as iron chelator is to burn down the residual stores of tumor tissue and, thus, to prevent iron organ disease from overload, another advantageous method which may be the removal of iron from the RBC membrane [9]. The decision for allogeneic marrow transplantation is generally considered appropriate only in patients with a complete HLA- donor (at least, 30- 40% of all patients). The decision to transfer patient to BMT is a frustrating issue with many ethical questions related to the length and quality of life of the transferred patient, compared to the patient being treated with BMT-supported care [10].
A closer examination of the problems of thalassemia patients treated with transplantation versus conventional treatment showed that the incidence of hypothyroidism, hypogonadism and cardiomyopathy was higher in patients treated with the convention method, while total sepsis and growth defects were higher in the transplanted patients. Iron overload should however be addressed in most BMT post patients, but can be treated with phlebotomy in patients with their hemoglobin level high enough to tolerate this type of treatment. Some patients may continue to require reduced doses of desferroxamine [7]. It is not possible to best treat the thalassemias with all its prevailing complications, unless stem cell transplant is carried out, which is uneconomical for the majority of population. So, the best way is to prevent the disease by educating people about prenatal and premarital screening [11].
Methodology
Study Design
A prospective cross-sectional study was carried out on the patients suffering from thalassemia. Patients experiencing thalassemia was chosen for the purpose of assessing the rate of prevalence, awareness, and prescription trends used by physicians. A questionnaire was prepared containing both open ended and closed ended questions. A consent form was prepared in English as well as Urdu to take permission of patient to participate in study.
Study Setting
Study was carried out in Sundas Foundation located in Sialkot, Pakistan. Permission was obtained from medical officer. Data was collected from General Medical Ward of Sundas Foundation located in Sialkot.
Duration of Study
The data was collected from August 2020-September 2020. Total period of data collection was almost 2 months.
Sample Size
Total number of thalassemia patients participated in this study was 70. Purpose of sample size is to make inferences about the population from a sample. Sample size effects precision of the evaluation of study as well as ability of the study to get the conclusions. Sample size represents the population to which the outcomes of study are to be generalized.
Patient Selection
The patients of any group were selected randomly from any regional variation, socio-economical society, demographic and age discrimination. Limitation of our research work in selection of patients includes that; patients were only being eligible if they were either resident of district Sialkot or its tehsil i.e. Daska, Sambrail and Pasroor. To reduce selection partiality within our research work, we report on all patients for whom there were treatment follow-up data, with no other criteria enforced.
Age and Gender
There is no restriction regarding age of patients for research work. Randomly 70 patients were collected of any age group to perform research. There is no constraint of religion or ethnicity in our research work. There was no discrimination of gender for prevalence of thalassemia patients, males and females both were included.
Patient Consent
The selected patients of thalassemia were agreed for their participation in our research project. Consent form was signed by patient in English and in Urdu for legal approaches.
Family History of Patient
Thalassemia is the genetic disorder passed down through families. Thalassemia develops in individuals with family history of either the diseased ones or carriers. Thalassemia is passed from parents to children through mutated hemoglobin genes. A child who inherits one mutated gene is carrier which is called thalassemia trait.
Study Tool
Well-structured data collection form or questionnaire was prepared, analyzed and then filled by in-person interview. All questions relevant to study including demographic characteristics (age, gender, socioeconomic status, and blood group), patient history (sign & symptoms, age of diagnosis, type of thalassemia, family history, complications (bone deformities, hepatitis, iron overload, infection, cardiovascular disorders, endocrine disorder) were included. Diagnostic tools for thalassemia include complete blood count (CBC), Hb- electrophoresis, renal function test, liver function test, thyroid function test, serum ferritin, calcium level and treatment protocols include pharmacological intervention and surgical intervention. Further pharmacological interventions include blood transfusion and other medications while surgical interventions include splenectomy and stem cell transplant.
Data Collection Procedure
The research was carried out in General medical ward of Sundas Foundation Sialkot. First of all, permission was obtained from medical officer of Sundas Foundation. 70 patients were selected randomly from hospital without any gender discrimination. All eligible patients and their parents were approached as they came in for routine follow-ups at thalassemia Centre. Written parental consent and child’s assent were obtained prior to participating in study. After identifying potential responder, consent was obtained from them. At the beginning of interview, all respondents were informed of the objectives of the study and were assured that all the information would remain confidential. Data was collected through preparing a structured questionnaire. All of the patients were allowed to answer the questionnaire concerning their medical history. Information was taken verbally from each participant and also from patient file. A questionnaire was designed for questioning about awareness of thalassemia ‟Do you know malnutrition can worsen thalassemia symptoms. Do you know about stem cell transplant? Do you know that blood transfusion is unhygienic conditions can lead to hepatitis? Do you know that frequent blood transfusion leads to iron overload?” The answers were helpful in accurate diagnosis of disease and treatment protocols (brand name, dose, frequency, dosage form).
Ethical Considerations
Data was collected after ethical approval of Sundas Foundation. A verbal consent was taken from each thalassemia patient before data collection.
Validity of Questionnaire
Face validity refers to the degree to which an evaluation, assessment or test subjectively appears to compute the variable or parameter that is supposed to measure. Face validity also called as logical validity. Content validity refers to extent to which the parameters of a test or evaluation are relevant to, and are fairly representative of entire domain; the test or evaluation seeks to measure.
Inclusion and Exclusion Criteria
The inclusion and exclusion criterion for patient selection is;
Inclusion Criteria Exclusion Criteria
Patients who signed the consent form and were interested in research work were included in this study. Patients outside the premises of Sialkot were excluded.
Patients diagnosed with thalassemia were included in this study. Patient without thalassemia were excluded.
Patients of both genders were included in this study. People with unethical believe were excluded.
Patients with any socioeconomic status were included. Patients suffering from any other blood disorder were excluded.
Data Analysis
The data was analyzed and interpreted after collection. The graphs and tables were plotted on Microsoft word and Microsoft excels to analyze the prevalence, level of awareness and prescription trends for thalassemia patients.
Results
Assessment of Awareness of Thalassemia
Sample size for this study was taken as 70 patients. In this study, data was collected from parents of patients suffering from thalassemia. Total 21 patients (30%) knew that thalassemia can be detected during pregnancy and 49 patients (70%) didn’t know about prenatal diagnosis. 58 patients (82.8%) were aware of the importance of nutrition in thalassemia and 12 patients (17.2%) were not aware of the value of nutrition in thalassemia. 34 patients (48.5%) knew about stem cell transplant and 36 patients (51.51%) didn’t know about this treatment. 48 patients (68.5%) were well aware that blood transfusion in unhygienic conditions can lead to hepatitis and 22 patients (31.5%) didn’t know this. 33 patients (47.1%) knew about the thalassemia trait and 37 patients (52.9%) had no idea about thalassemia trait. 58 patients (82.8%) knew that thalassemia requires the lifelong treatment and 12 patients (17.2%) didn’t know that. 60 patients (85.7%) were well aware that thalassemia patients can lead a healthy life with proper treatment and 10 patients (14%) didn’t know that. 62 patients (88.5%) knew about iron overload and 8 patients (11.5%) have no idea about iron overload. 48 patients (68.5%) knew that thalassemia is a genetic disorder and 22 patients (31.5%) didn’t know that. 53 patients (75.7%) knew that every thalassemia patient doesn’t require blood transfusion and 17 patients ( 24.3%) didn’t know that (Table 1 & Figure 1).
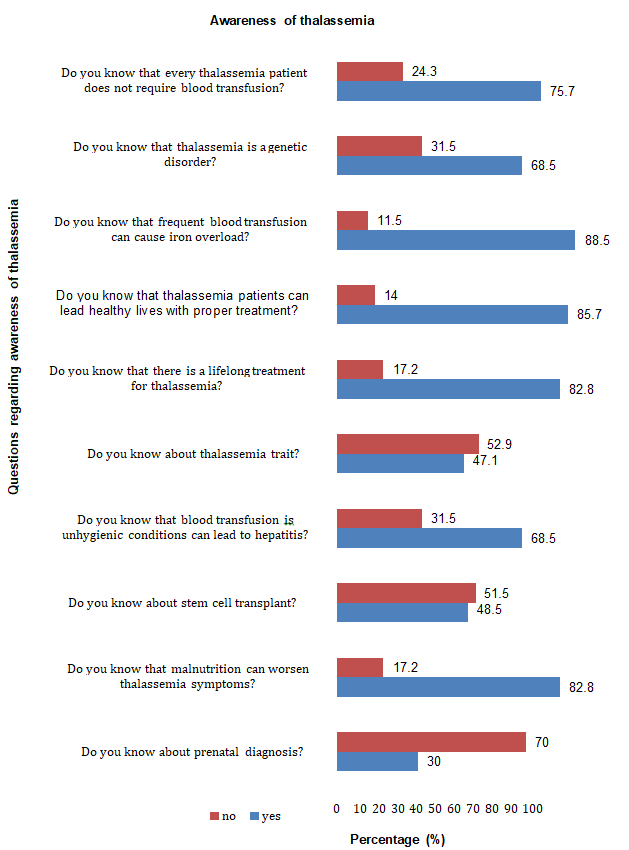
| Sr. No. | Questions | Yes | No | ||
|---|---|---|---|---|---|
| Frequency (n) | Percentage (%) | Frequency (n) | Percentage (%) | ||
| 1 | Do you know about prenatal diagnosis? | 21 | 30 | 49 | 70 |
| 2 | Do you know that malnutrition can worsen thalassemia symptoms? | 58 | 82.8 | 17.2 | |
| 3 | Do you know about stem cell transplant? | 34 | 48.5 | 36 | 51.5 |
| 4 | Do you know that blood transfusion is unhygienic conditions can lead to hepatitis? | 48 | 68.5 | 22 | 31.5 |
| 5 | Do you know about thalassemia trait? | 33 | 47.1 | 37 | 52.9 |
| 6 | Do you know that there is a lifelong treatment for thalassemia? | 58 | 82.8 | 12 | 17.2 |
| 7 | Do you know that thalassemia patients can lead healthy lives with proper treatment? | 60 | 85.7 | 10 | 14 |
| 8 | Do you know that frequent blood transfusion can cause iron overload? | 62 | 88.5 | 8 | 11.5 |
| 9 | Do you know that thalassemia is a genetic disorder? | 48 | 68.5 | 22 | 31.5 |
| 10 | Do you know that every thalassemia patient does not require blood transfusion? | 53 | 75.7 | 17 | 24.3 |
Table 1: Assessment of awareness of thalassemia.
Assessment of Prevalence of Thalassemia
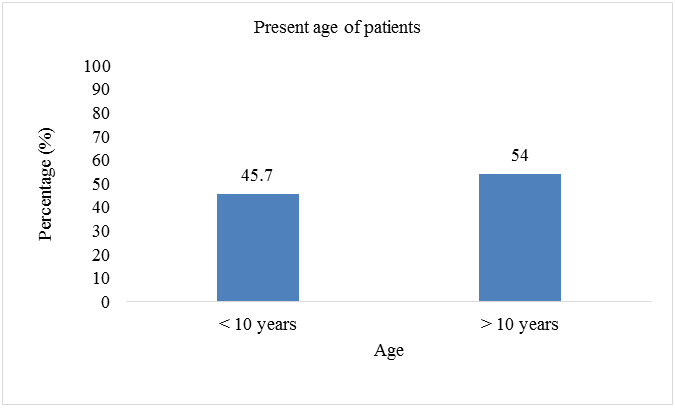
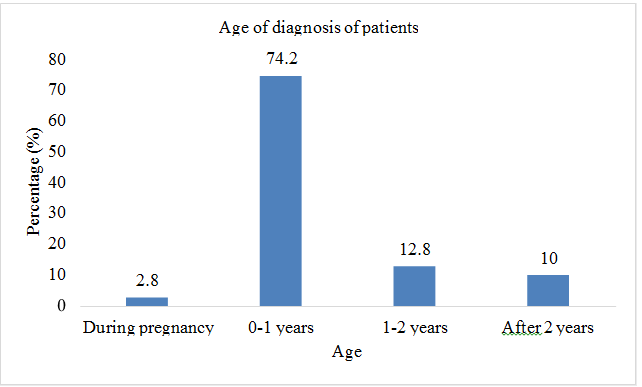
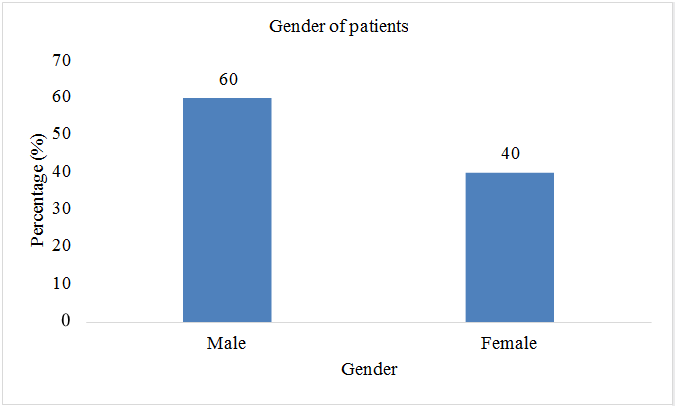
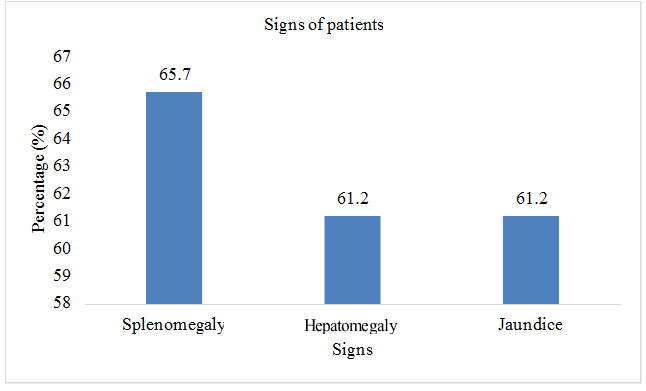
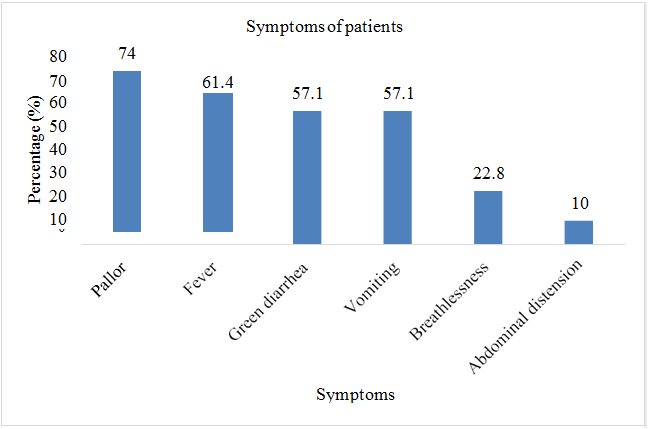
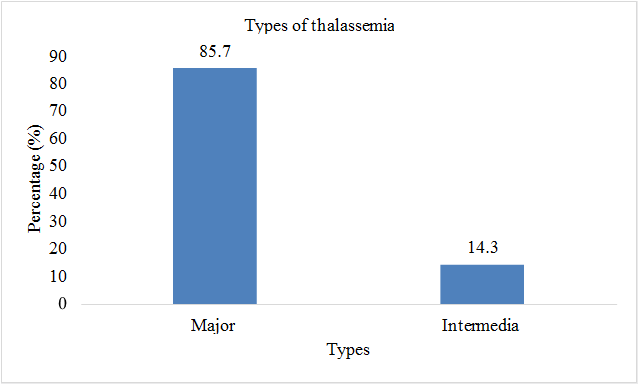
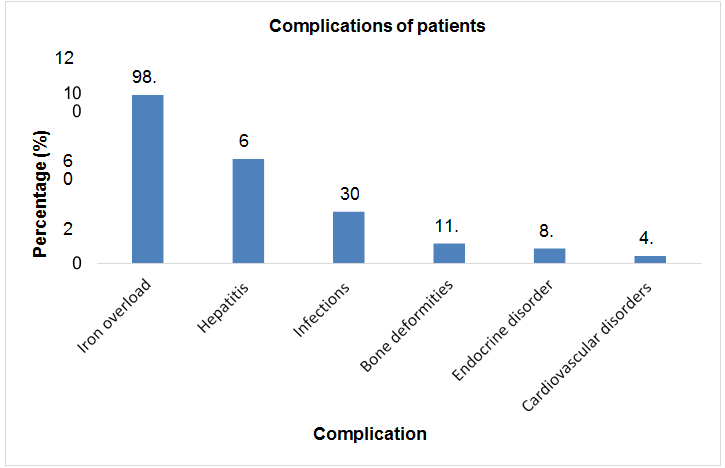
Total sample size was taken as 70 patients. Present age of patients (n=32; %=45.7) was less than 10 years and (n=38;%=54.3) had present age greater than 10. Patients (n=2; %=2.8) were diagnosed with thalssemia during pregnancy, (n=52; %=74.2) were diagnosed at the age of 0-1 year, (n=9; %=12.8) were diagnosed at the age of 1-2 year, and (n=7; %=10) were diagnosed after two years of birth. Out of 70 patients involved in this study, (n=42; %=60) were males, and (n=28; %=40) were females. Patients (n=43; %=61.2) showed the sign of hepatomegaly, (n=46; %=65.7) showed the sign of splenomegaly, and (n=43; %=61.2) showed the sign of jaundice. Patients (n=16; %=22.8) had the symptom of breathlessness, (n=7; %=10) had abdominal distention, (n=43; %=61.4) had fever as initial symptom, (n=40; %=57.1) had green diarrhea, (n=40; %=57.1) had vomiting, patients (n=52; %=74) had pallor. All 70 patients (100%) had the family history of thalassemia minor in both parents. Out of 70, 60 patients (85.7%) had major thalssemia and 10 (14.3%) had thalassemia intermedia. (n=69; %=98.5) had iron overload as a complication of blood transfusion, (n=43; %=61) had hepatitis, (n=21; %=30) had infections, (n=8; %=11.4) had bone deformities, (n=6; %=8.5) had endocrine disorders, (n=3; %=4.2) had cardiovascular complications (Table 2 & Figures 2-8).
| Parameters | Frequency (n) | Percentage (%) |
|---|---|---|
| Present age | ||
| <10 years | 32 | 45.7 |
| > 10 years | 38 | 54.3 |
| Age of diagnosis | ||
| During pregnancy | 2 | 2.8 |
| 0-1 years | 52 | 74.2 |
| 1-2 years | 9 | 12.8 |
| After 2 years | 7 | 10 |
| Gender | ||
| Male | 42 | 60 |
| Female | 28 | 40 |
| Signs | ||
| Splenomegaly | 46 | 65.7 |
| Hepatomegaly | 43 | 61.2 |
| Jaundice | 43 | 61.2 |
| Symptoms | ||
| Pallor | 52 | 74 |
| Fever | 43 | 61.4 |
| Green diarrhea | 40 | 57.1 |
| Vomiting | 40 | 57.1 |
| Breathlessness | 16 | 22.8 |
| Abdominal distension | 7 | 10 |
| Family history | ||
| Maternal | 0 | 0 |
| Paternal | 0 | 0 |
| Carrier (both) | 70 | 100 |
| Types of thalassemia | ||
| Major | 60 | 85.7 |
| Intermedia | 10 | 14.3 |
| Complications | ||
| Iron overload | 69 | 98.5 |
| Hepatitis | 43 | 61 |
| Infections | 21 | 30 |
| Bone deformities | 8 | 11.4 |
| Endocrine disorder | 6 | 8.5 |
| Cardiovascular disorders | 3 | 4.2 |
Table 2: Assessment of prevalence of thalassemia.
Assessment of the Diagnostic Tools of Thalassemia
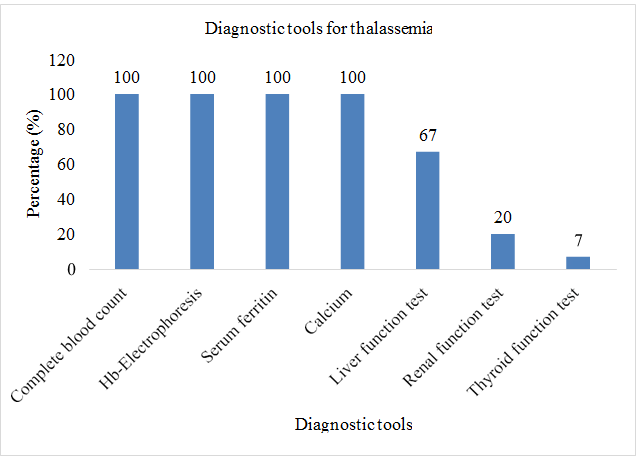
Sample size for this study was taken as 70. Complete blood count test was performed on patients (n=70; %=100). Hb- electrophoresis was performed on patients (n=70; %=100). Serum ferritin level test was carried out for patients (n=70; %=100). Serum calcium level test was performed on (n=70; %=100). Liver function test was carried out for patients (n=47; %=67). Renal function test was conducted on (n=14; %=20). Thyroid function test was performed on patients (n=5; %=71) (Table 3 & Figure 9).
| S. No. | Laboratory tests | Frequency (n) | Percentage (%) |
|---|---|---|---|
| 1 | Complete blood count | 70 | 100 |
| 2 | Hb-Electrophoresis | 70 | 100 |
| 3 | Serum ferritin | 70 | 100 |
| 4 | Calcium | 70 | 100 |
| 5 | Liver function test | 47 | 67 |
| 6 | Renal function test | 14 | 20 |
| 7 | Thyroid function test | 5 | 7 |
Table 3: Assessment of diagnostic tools of thalassemia.
Assessment of Treatment Trends of Thalassemia
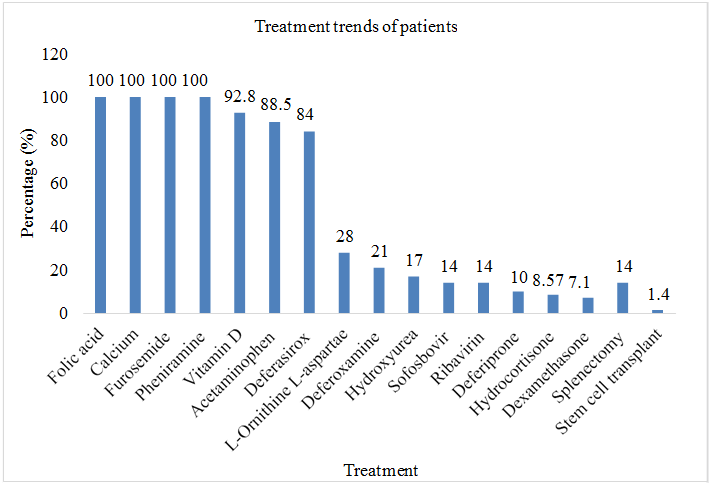
Total sample size was taken as 70 patients. Patients, (n=70; %=100) had received furosemide, pheniramine before transfusion, (n=9; %=12.8) had received ceftriaxone, (n=6; %=8.57) had received hydrocortisone, (n=5; %=7.1) had received hydrocortisone before blood transfusion. All 70 patients were taking folic acid and calcium on daily basis. Patients (n=65; %=92.8) were regularly receiving vitamin D. Patients (n=59; %=84) were taking deferasirox as iron chelating therapy, (n=15; %=21) were receiving deferoxamine as iron chelating therapy, (n=15; %=21) were receiving hydroxyurea as iron chelating therapy, (n=12; %=17) were taking deferiprone as iron chelating therapy. Patients (n=10; %=14) were receiving sofosbovir and ribavirin as a treatment of HCV, (n=20; %=28) were taking L-Ornithine L-Aspartate as a treatment for HCV. Out of 70 patients, 10 (14.2%) had undergone through splenectomy and only 1 patient (1.4%) had undergone through stem cell transplant (Table 4 & Figure 10).
| Medicines | Frequency (n=70) | Percentage (%) |
|---|---|---|
| Pharmacological Interventions | ||
| Folic acid | 70 | 100 |
| Calcium | 70 | 100 |
| Furosemide | 70 | 100 |
| Pheniramine | 70 | 100 |
| Vitamin D | 65 | 92.8 |
| Acetaminophen | 62 | 88.5 |
| Deferasirox | 59 | 84 |
| L-Ornithine L- | 20 | 28 |
| Aspartate | ||
| Deferoxamine | 15 | 21 |
| Hydroxyurea | 12 | 17 |
| Sofosbovir | 10 | 14 |
| Ribavirin | 10 | 14 |
| Deferiprone | 7 | 10 |
| Hydrocortisone | 6 | 8.57 |
| Dexamethasone | 5 | 7.1 |
| Surgical interventions | ||
| Splenectomy | 10 | 14 |
| Stem cell transplant | 1 | 1.4 |
Table 4: Assessment of treatment trends of thalassemia.
Discussion
Thalassemia is the most common genetic disorder among the people of Pakistan. This is probably due to the lack of knowledge about the pre-marital and prenatal diagnosis of thalassemia. Majority (n=49; %=70) of the parents of patients suffering from thalassemia included in this study didn’t know about prenatal diagnosis. Results of this study are relatable to the study performed in Rahim Yar khan in 2016 which also shows that most of parents of thalassemia patients were unaware about the prenatal diagnosis [12].
Patients suffering from thalassemia major require frequent blood transfusion, which results in the accumulation of iron in various body organs leading to the damage of liver, heart, endocrine glands, and kidneys. Majority (n=62; %=88.5) of the participants involved in this study knew about the complication of iron overload. Results of this study are comparable to the study conducted in Peshawar in 2015 which depicts the high level of awareness among the parents about iron overload [13].
Life expectancy of patients of thalassemia has been increased in past few years due to the regular use of blood transfusion and iron chelation therapy. In developing countries, Government and various NGOs have provided the facilities for transfusions and iron chelation therapy free of cost for the patients who cannot afford this. This has increased the life expectancy of thalassemia patients in developing countries. In our study, (n=38; %=54.3) were aged more than 10 years. Results of this study are relatable to the study performed in London in 2009 which shows that life expectancy of thalassemia patients has increased above 10 years [14].
Thalassemia in majority of patients (n=52; %=74.2) had diagnosed within one year of age. Results of this study are relatable to the study conducted in Bangladesh in 2018 [15]. Majority of the patients (n=42; %=60) enrolled in this study were male, which depicts the greater prevalence of thalassemia in males as compared to females because parents give more attention to their male child and ready to spend more money on male child compared to female ones. Furthermore, investigations have shown that thalassemia is a single gene disorder inherited by the recessive mean of transmission. Results of this study are comparable to the study performed in India in 2015 [16]. Majority of patients (n=43; %=61.2) had hepatomegaly, patients (n=46; %=65.7) had splenomegaly, which means that hepatomegaly and splenomegaly are the major signs of thalassemia. These results are relatable to the study performed in Rawalpindi in 2004 [17]. In our study, majority of patients (n=52; %=74) had the symptoms of pallor, patients (n=43; %=61.4) had the symptoms of fever. Results of this study are relatable to the study performed in Bangladesh in 2018 [15]. Patients suffering from thalassemia major have the critical form of the disease and need chronic transfusion. A thalassemia inter-medium is the form of thalassemia featured by the less critical symptoms as compared to thalassemia major. Majority of the patients (n=60; %=85.7) were suffering from thalassemia major and only (n=10; %=14.3) were suffering from thalassemia intermedia. Results of this study are comparable to the study conducted in Jammu and Kashmir in 2014 [18].
Patients suffering from thalassemia have some severe transfusion related complications; most common among those is iron overload and infection. In our study, majority (n=69; %=98.5) of the patients had increased serum ferritin level. Results of this study are relatable to the study performed in Italy in 2017 [19]. Most common infection related to blood transfusion in thalassemia is hepatitis. In our study second most common complication reported is hepatitis, (n=43, %=61) patients were tested positive for hepatitis. Results of this study are comparable to the study performed in Islamabad, Pakistan in 2014 [20].
In our study, all cases were diagnosed by the hemoglobin electrophoresis (n=70; %=100). This assessment is related to the study performed in India in 2018 [21]. For the measurement of the occurrence and level of complications, all the patients (n=70; %=100) underwent CBC with differential and serum ferritin level assessment on routine basis. This result is relatable to the study performed in North America for the purpose of establishing the standard guidelines for the monitoring of the patients suffering from thalassemia [22].
Patients suffering from the thalassemia major require repeated blood transfusions. Blood transfusion may leads to the transfusion associated circulatory overload. This can be prevented by the use of diuretics before transfusion. Furosemide is the most effective diuretic used as a premedication in blood transfusion. In our study, all the patients (n=70; %=100) were receiving furosemide intravenously before transfusion. The result is consistent with the study conducted in Canada in 2013 [23]. Malnutrition is quite frequent in thalassemia patients. So, they regularly require dietary supplements to maintain their health. Most commonly prescribed dietary supplements according to our study were; folic acid (n=70; %=100), calcium (n=70; %=100), vitamin D (n=65; %=92.8). This result is relatable to the study performed in United States and Canada in 2012 [24]. Thalassemia patients receive iron chelation therapy in order to reduce the iron accumulation in body resulted from repeated blood transfusion. In our study most commonly used iron chelator was defesirox (n=59; %=84) and deferoxamine (n=15; %=21). These results are consistent with the results of the study conducted in Turkey [25].
In thalassemia, there is an increase destruction of RBCs which results in hypersplenism. Splenomegaly results in an increase in blood transfusion requirement. Physicians recommend the splenectomy when the need for blood transfusion exceeds 220mL RBCs/kg/y. In our study (n=10; %=14) patients had undergone through splenectomy. This result is comparable to the study performed in Iran in 2010 [26].
The ultimate treatment for thalassemia is stem cell transplant. This treatment is offered by the limited number of hospitals in Pakistan. The average cost for bone marrow transplant is unaffordable for the majority of our population. In our study only (n=1; %=1.4) patient had undergone through this treatment. This result is comparable to the study in Karachi, Pakistan [27].
Conclusion
Thalassemia is amongst the common genetic blood disorders. According to our study, awareness among the parents regarding thalassemia was insufficient. Hb- electrophoresis is used as conformational diagnostic tool for thalassemia. Treatment of thalassemia is associated with a lot of complications. To assess the complications, CBC, serum ferritin, LFT, RFT and serum calcium monitoring must be carried out regularly for all the patients. Patients suffering from thalassemia regularly require blood transfusion, which results in iron overload in body. Defesirox is the most commonly used iron chelator for treating iron overload. Ultimate treatment is stem cell transplant which is out of the reach of most of population. So, best way is to prevent thalassemia by enlightening the importance of pre-natal and pre-marital diagnosis of thalassemia.
Recommendations
Prime concentration should be laid towards the obviation of disease, which requires the enforcement by Government officials for premarital diagnosis. Proper campaign must be directed for educating the general public about the complexity of thalassemia.
Limitation of Study
Primary limitation of this study is that, it includes only one thalassemia patient who had undergone through the stem cell transplant, so this study does not provide a complete picture of the assessment of this treatment. This is due to the fact that this study is restricted in NGO sector of the single district, where majority of the patients can’t afford this expensive treatment.
Authors’ Contributions
Faiza Naeem proposed the idea of this project and designed the study. Hazima Tariq and Kainat performed the survey for data collection. Nimra Riasat, saba Zulfiqar and sidra mubarak compiled the data. Faiza Naeem analyzed the data. All authors equally contributed and wrote the manuscript. Faiza Naeem coordinated the project preparation of the final version of the manuscript.
Acknowledgment
The study was conducted by the Islam College of Pharmacy, Sialkot.
References
-
Muncie HL, Campbell JS (2009) Alpha and beta thalassemia. American family physician 80(4): 339-344.
-
Cao A, Kan YW (2013) The prevention of thalassemia. Cold Spring Harbor perspectives in medicine 3(2): a011775.
-
Delea TE, Edelsberg J, Sofrygin O, Thomas SK, Baladi JF, et al. (2007) Consequences and costs of noncompliance with iron chelation therapy in patients with transfusion‐ dependent thalassemia: a literature review. Transfusion 47(10): 1919-1929.
-
Ahmed S, Saleem M, Modell B, Petrou M (2002) Screening extended families for genetic hemoglobin disorders in Pakistan. New England Journal of medicine 347(15): 1162-1168.
-
Shang X, Xu X (2017) Update in the genetics of thalassemia: What clinicians need to know. Best Practice & Research Clinical Obstetrics & Gynaecology 39: 3-15.
-
Baig SM, Azhar A, Hassan H, Baig JM, Aslam M, et al. (2006) Prenatal diagnosis of β‐ thalassemia in Southern Punjab, Pakistan. Prenatal diagnosis 26(10): 903-905.
-
Borgna Pignatti C, Rugolotto S, Stefano PF, Zhao H, Cappellini MD, et al. (2004) Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica 89(10): 1187-1193.
-
Borgna-Pignatti C, Rugolotto S, De Stefano P, Piga A, Di Gregorio F, et al. (1998) Survival and disease complications in thalassemia major. Ann N Y Acad Sci 850(1): 227-231.
-
Rund D, Rachmilewitz E (2000) New trends in the treatment of β-thalassemia. Critical reviews in oncology/ hematology 33(2): 105-118.
-
Rund D, Rachmilewitz E (1995) Advances in the pathophysiology and treatment of thalassemia. Critical reviews in oncology hematology 20(3): 237-254.
-
Arif F, Fayyaz J, Hamid A (2008) Awareness among parents of children with thalassemia major. J Pak Med Assoc 58(11): 621-624.
-
Ghafoor MB (2016) Level of awareness about thalassemia among parents of thalassaemic children. Journal of Rawalpindi Medical College 20(3): 209-211.
-
Ali S, Saffiullah MF (2015) Awareness of parents regarding beta thalassemia major disease. Khyber Med Univ J 7(2): 72-75.
-
Telfer P (2009) Update on survival in thalassemia major. Hemoglobin 33(S 1): S76-S80.
-
Ali MR, Bari MI, Mia MSH, Rahman MK., Hossain MF, et al. (2018) Clinical Profile of Thalassemia Syndrome in Children of Northern Bangladesh. TAJ Journal of Teachers Association 31(2): 6-11.
-
Bhatia P, Nagar V, Meena JS, Singh D, Pal DK (2015) A study on the demographic and morbidity patterns of thalassemia patients registered at a tertiary-care center of central India. Int J Med Sci Public Health 4(1): 85-88.
-
Younus M, Hassan K, Ikram N, Naseem L, Zaheer HA, et al. (2004) Hepatitis C virus seropositivity in repeatedly transfused thalassemia major patients. Int J Pathol 2(1): 20-23.
-
Vasudev R, Sawhney V (2014) Thalassemia major and intermedia in Jammu and Kashmir, India: A regional transfusion centre experience. Indian Journal of Hematology and Blood Transfusion 30(4): 297-300.
-
Bonifazi F, Conte R, Baiardi P, Bonifazi D, Felisi M, et al. (2017) Pattern of complications and burden of disease in patients affected by beta thalassemia major. Current Medical Research and Opinion 33(8): 1525-1533.
-
Din G, Malik S, Ali I, Ahmed S, Dasti JI (2014) Prevalence of hepatitis C virus infection among thalassemia patients: a perspective from a multi-ethnic population of Pakistan. Asian Pacific journal of tropical medicine 7: S127-S133.
-
Joseph N, Pai S, Sengupta S, Bharadwaj S, Dhawan S, et al. (2018) A clinico-epidemiological study of thalassemia cases in India. Journal of Natural Science Biology and Medicine 9(2): 236.
-
Tubman VN, Fung EB, Vogiatzi M, Thompson AA, Rogers ZR, et al. (2015) Guidelines for the standard monitoring of patients with thalassemia: report of the thalassemia longitudinal cohort. Journal of pediatric hematology oncology 37(3): 162-169.
-
Alam A, Lin Y, Lima A, Hansen M, Callum JL (2013) The prevention of transfusion-associated circulatory overload. Transfusion Medicine reviews 27(2): 105-112.
-
Fung EB, Xu Y, Trachtenberg F, Odame I, Kwiatkowski JL, et al. (2012) Inadequate dietary intake in patients with thalassemia. Journal of the Academy of Nutrition and Dietetics 112(7): 980-990.
-
Yaman A, Pamir ISIK, Yarali N, Karademir S, Cetinkaya S, et al. (2013) Common complications in beta-thalassemia patients. International Journal of Hematology and Oncology 29(4): 193-199.
-
Mahyar A, Ayazi P, Pahlevan AA, Mojabi H, Sehhat MR, et al. (2010) Zinc and copper status in children with Beta- thalassemia major. Iranian journal of pediatrics 20(3): 297.
-
Shamsi TS, Hashmi K, Adil S, Ahmad P, Irfan M, Raza S, et al. (2008) The stem cell transplant program in Pakistan- the first decade. Bone marrow transplantation 42(1): S114-S117.
- hMPV: Is It Another Covid-19 Like Situation?
- Streptomyces: Sources of Novel Discoveries in Antibiotic Research to Combat Antimicrobial Resistance
- A Review of Mosquitoes (Diptera: Culicidae) and Their Biodiversity, Medical and Veterinary Importance
- Past and Current Immunotherapy in Cancer
- Hematological Cancer and Viral Infection
- The Growing Threat of Antimicrobial Resistance in India: Challenges and Solutions