Varstation: a Complete and Efficient Tool to Support NGS Data Analysis
Varstation is a cloud-based NGS data processor and analyzer for human genetic variation. This resource provides a customizable, centralized, safe, and clinically validated environment aiming to improve and optimize the flow of NGS analyses and reports related with clinical and research genetics.
Introduction
In recent years, advances in next-generation sequencing (NGS) technologies and applications, such as whole-genome sequencing (WGS), whole-exome sequencing (WES), and targeted-sequencing have enabled the generation of large- scale data and identification of millions of genetic variants both for research and clinical diagnostic purposes Yang Y, et al [1]; Xue Y, et al. [2]. Identifying, interpreting, classifying, and associating human genetics variants with phenotypic particularities among individuals or diseases are important parts of the process to accomplish personalized medicine Gaspar P, et al. [3]; Geoffroy V, et al. [4].
Data workflow in NGS includes several bioinformatics steps from the analysis of raw sequencing data transforming the signal from the sequencers to raw sequences that are further aligned and compared with the reference genome Geoffroy V, et al. [4]. In general, a typical workflow analysis consists of the following steps: raw data quality control (QC), preprocessing, alignment, post-alignment processing, variant calling, annotation, and prioritization Bao R, et al. [5].
There is a pressing need for the development of optimized workflows and variant analysis toolkits that assist clinical and laboratory geneticists and researchers in identifying, classifying and ranking variants in a timely fashion.
We describe a new powerful tool named Varstation (Figure 1) to support the human NGS data analysis workflows. Varstation is a cloud-based solution for computational processing and clinical support of NGS genetic testing that provides a customizable, centralized, safe, and clinically validated environment. This resource was developed by a multidisciplinary team composed by bioinformaticians, biologists, geneticists, software engineers, and designers. The goal was to design a resource that could be used for different analysis workflows being both scientifically sound and easy to use.
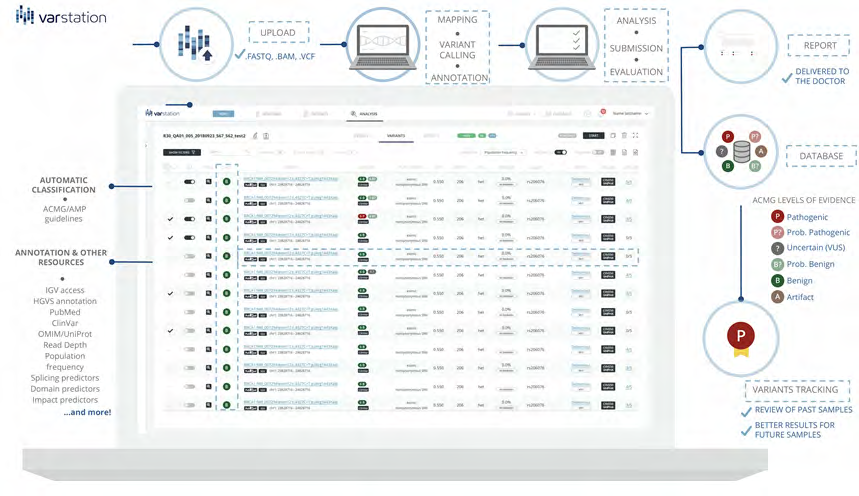
Software Description
The Varstation
Varstation comprises three main components of bioinformatics pipelines: alignment of the sequenced readings to the reference genome; variant calling and annotation, which entails capturing and summarizing all existing information about the variant across multiple public databases.
The strength of Varstation workflow is based on:
- Flexibility: files from any DNA sequencing platform, in any format (.fastq, .bam, and .vcf) are processed to generate results.
- End-to-end and automated processing: evaluation of quality parameters, mapping, multiple variant callers, database annotation and automatic variant pre-classification according recommendations and guidelines of the American College of Medical Genetics and Genomics (ACMG), the Association of Molecular Pathology (AMP), the College of American Pathologists (CAP) (Richards et al., 2015) and the Clinical Laboratory Accreditation Program (Programa de Acreditação de Laboratórios Clínicos, PALC) (http://www.sbpc.org.br/ programas-da-qualidade/documentos-do-palc/).
- Support for clinical interpretation: more than 100 genetic mutations databases are incorporated, including data from germline, somatic and structural variants.
- Robust filters: filtering engine based on all annotated mutation data.
- Clear and structured results: relevant clinical information to support the medical report. In addition, Varstation provides visualization of data features to share results with other institutions. Information from the external databases and filters provides in Varstation are described in supplementary table S1.
Workflow Optimization: Main Features
To access Varstation, the user must register using a valid e-mail address. After an e-mail confirmation, the new user will be able to access the platform and the new users onboarding tutorial (raw clinical exome data sample are available). Users can update and personalize the workflow depending on the type of the starting file and adjust parameters each component.
QC of raw data and their preprocessing can be performed using the Fast QC Winget SW, et al. [6], BED Tools Quinlan AR, et al. [7], Bam Tools Barnett DW, et al. [8] and VCF tools Danecek P, et al. [9] toolkits. After raw data QC and preprocessing, the next step is to map readings to the reference genome and process the variant calling with high efficiency and accuracy. Two of the main mapping and alignment tools are available in the platform: Burrows- Wheeler Aligner (BWA) Li H, et al. [10] and Torrent Mapping Alignment Program TMAP (https://github.com/iontorrent/ TS/tree/master/Analysis/TMAP).
Programs available for germline variant calling include GATK-Unified Genotyper and Haplotype Caller Van der Auwera GA, et al. [11], SAM tools Li H, et al. [12], Free Bayes Garrison E, et al. [13], Atlas Challis D, et al. [14] and smCounter Xu C, et al. [15]. For somatic variant detection, Varstation provides GATK Van der Auwera GA, et al. [11], SAM tools Li H, et al. [12], VarScan Koboldt DC, et al. [16], Lai Z, et al. [17] and smCounter Xu C, et al. [15].
After variant identification, the annotation attributes such as genomic components, gene symbol, amino acid change, and functional consequences are attached to the variant list. For annotation, Varstation offers one in-house annotation algorithm that was based on ANNOVAR, the most commonly used variant annotation programs Wang K, et al. [18].
The last step is variant filtration and prioritization. Users can filter the variants by gene name, ACMG Richards S, et al. [19] or ClinVar Landrum MJ, et al. [20] classification, protein coding consequence, variant allele frequency (VAF), depth coverage, frequency in public databases, specifically: 1000g, ESP6500, gnomAD, ABraOM, and ExAC 1000 Genomes Project Consortium, et al. [21]; Tennessen JA, et al. [22]; Karczewski KJ, et al. [23]; Naslavsky MS, et al. [24]; Lek M, et al. [25], presence in dbNSP Sherry ST, et al. [26], protein- damage prediction tools (VEST, FATHMM, SIFT, Polyphen, PROVEAN, CADD) Douville C, et al. [247]; Carter H, et al. [28]; Shihab HA, et al. [29]; Shihab HA, et al. [30]; Shihab HA, et al. [31]; Sim NL, et al. [32]; Adzhubei I, et al. [33]; Choi Y, et al. [34]; Rentzsch P, et al. [35], disease associated by OMIM Rentzsch P, et al. [36] and UniProt Consortium [37]. Variants are also linked to any associated phenotypes in the Human Phenotype Ontology (HPO) Köhler S, et al. [38]. Users can also easily visualize the variant of interest, in their context of the .bam files, using the Integrative Genomics Viewer Robinson JT, et al. [39].
One of the key features of Varstation is that users can build their organization’s internal database, customizing their own filter’s pipelines, link variants to phenotypes, diseases or articles, and can make their own pathogenicity assessments, potentially allowing for the rapid identification of recurrent mutations, and periodic variant reevaluation or reanalysis, following the ACMG guidelines Deigna JL, et al. [40]. This feature is extremely relevant mainly for clinical laboratories that need policies and protocols established for variant re-analyzes. Besides that, Varstation also provides multiple ready-to-use standardized pipelines.
Finally, Varstation enables the analysis of each sample by up to 3 experts to guarantee accuracy of results, and then it is possible to generate a customized report with a list of candidate variants that will be reported.
Conclusion
The process of analyzing NGS data in Varstation improves performance, traceability, and safety of analyses; in addition the resource complies with the good international practices for analysis of human genetic variants.
Varstation is a complete, simple, and powerful tool to process and analyze NGS data that has already been adopted by leading laboratories in Brazil. The initial project of Varstation was developed in June 2015 and became available for use only for the Sociedade Beneficiente Hospital Israelita Albert Einstein hospital and the Genomika laboratory. In 2018, Varstation also became available to other laboratories, hospitals, and research centers. Since then, Varstation has being used by approximately 500 organizations and has processed more than 10,000 samples.
As one of Varstation best features, the software has an online chat service that helps users to solve problems found during the analysis process. In addition, a multi-disciplinary team performs hands-on courses to help the community to further improve the quality of their genomic analysis.
Conflicts of Interest: None Declared.
Acknowledgements
We would like to thank all colleagues from the Clinical Laboratory of Hospital Israelita Albert Einstein for the partnership and the enriching discussions. We would like to also thank to all developers from the Innovation Laboratory that did contribute and keep working in Varstation, offering an incredible atmosphere of work.
References
-
Yang Y, Muzny DM, Xia F, Niu Z, Person R, et al. (2014) Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312(18): 1870-1879.
-
Xue Y, Ankala A, Wilcox WR, Hegde MR (2015) Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: single-gene, gene panel, or exome/genome sequencing. Genet Med 17(6): 444-451.
-
Gaspar P, Lopes P, Oliveira J, Santos R, Dalgleish R (2014) Variobox: automatic detection and annotation of human genetic variants. Hum Mutat 35(2): 202-207.
-
Geoffroy V, Pizot C, Redin C, Piton A, Vasli N, et al. (2015) VaRank: a simple and powerful tool for ranking genetic variants. PeerJ 3: e796.
-
Bao R, Huang L, Andrade J, Tan W, Kibbe WA, et al. (2014) Review of current methods, applications, and data management for the bioinformatics analysis of whole exome sequencing. Cancer Inform 13(S 2): 67-82.
-
Winget SW, Andrews S (2018) FastQ Screen: A tool for multi-genome mapping and quality control. F1000Res 7: 1338.
-
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26(6): 841-842.
-
Barnett DW, Garrison EK, Quinlan AR, Strömberg MP, Marth GT (2011) BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27(12): 1691-1692.
-
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, et al. (2011) The variant call format and VCFtools. Bioinformatics 27(15): 2156-2158.
-
Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv.
-
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, et al. (2013) From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43(1110).
-
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16): 2078-2079.
-
Garrison E, Marth G (2012) Haplotype-based variant detection from short-read sequencing. arXiv.
-
Challis D, Yu J, Evani US, Jackson AR, Paithankar S, et al. (2012) An integrative variant analysis suite for whole exome next-generation sequencing data. BMC Bioinformatics 13: 8.
-
Xu C, Gu X, Padmanabhan R, Wu Z, Peng Q, et al. (2018) smCounter2: an accurate low-frequency variant caller for targeted sequencing data with unique molecular identifiers. Bioinformatics 35(8): 1299-1309.
-
Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, et al. (2012) VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22: 568-576.
-
Lai Z, Markovets A, Ahdesmaki M, Chapman B, Hofmann O, et al. (2016) VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res 44(11): e108.
-
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high- throughput sequencing data. Nucleic Acids Res 38(16): e164.
-
Richards S, Aziz N, Bale S, Bick D, Das S, et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5): 405-424.
-
Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, et al. (2018) ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 46(D1): D1062-D1067.
-
1000 Genomes Project Consortium, Abecasis GR, Altshuler D, Auton A, Brooks LD, et al. (2010) A map of human genome variation from population-scale sequencing. Nature 467(7319): 1061-1073.
-
Tennessen JA, Bigham AW, O’connor TD, Fu W, Kenny EE, et al. (2012) Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337(6090): 64-69.
-
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, et al. (2019) Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. BioRxiv.
-
Naslavsky MS, Yamamoto GL, de Almeida TF, Ezquina SAM, Sunaga DY, et al. (2017) Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum Mutat 38(7): 751-763.
-
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536(7616): 285-291.
-
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, et al. (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29(1): 308-311.
-
Douville C, Masica DL, Stenson PD, Cooper DN, Gygax DM, et al. (2016) Assessing the Pathogenicity of Insertion and Deletion Variants with the Variant Effect Scoring Tool (VEST-Indel). Hum Mutat 37(1): 28-35.
-
Carter H, Douville C, Stenson PD, Cooper DN, Karchin R (2013) Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genomics 14(S3): S3.
-
Shihab HA, Gough J, Mort M, Cooper DN, Day INM, et al. (2014) Ranking non-synonymous single nucleotide polymorphisms based on disease concepts. Hum Genomics 8(1): 11.
-
Shihab HA, Gough J, Cooper DN, Day INM, Gaunt TR, et al. (2013) Predicting the functional consequences of cancer- associated amino acid substitutions. Bioinformatics 29(12): 1504-1510.
-
Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GLA, et al. (2013) Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat 34(1): 57-65.
-
Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC (2012) SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res 40: W452-W457.
-
Adzhubei I, Jordan DM, Sunyaev SR (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet, Chapter 7.
-
Choi Y, Chan AP (2015) PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31(16): 2745-2747.
-
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M, et al. (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47: D886-D894.
-
McKusick VA (2007) Mendelian Inheritance in Man and its online version, OMIM. Am J Hum Genet 80(4): 588- 604.
-
UniProt Consortium (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47(D1): D506-D515.
-
Köhler S, Carmody L, Vasilevsky N, Jacobsen JOB, Danis D, et al. (2019) Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res 47(D1): D1018-D1027.
-
Robinson JT, Thorvaldsdóttir H, Wenger AM, Zehir A, Mesirov JP (2017) Variant Review with the Integrative Genomics Viewer. Cancer Res 77(21): e31-e34.
-
Deigna JL, Chung WK, Kearney HM, Monaghan KG, Rehder CW, et al. (2019) Points to consider in the reevaluation and reanalysis of genomic test results: a statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med 21: 1267-1270.
- Carbon Code for Analysis of Protein Stability in Protein Mutation
- Number of Contiguous Amino Acids in Nanon of 16A Diameter
- Identification of Hub Genes and Pathways in Cervical Cancer by Statistical and Bioinformatics Analysis
- Effect of Dietary Inclusion Levels of Moringa Olerifera Oil on the Growth Performance and Nutrient Retention of Broiler Starter Chicks
- Proteomics Loans in Kinetoplastids during the Last Decade
- “Identification of SARS-CoV-2 in Human Genome based on Protein Dynamics Conversion and Target Genes Marking via Bioinformatics Approaches”