Superposition of Cryo-EM and AlphaFold Predictions of Dengue Antigen-Antibody Complexes
Accurate prediction of antibody-antigen interfaces is essential for structure-based drug design (SBDD) an vaccine development. The dengue virus envelope (E) protein is a major target of neutralizing antibodies and has been extensively characterized structurally. Although the deep-learning methods such as AlphaFold has achieved near-experimental accuracy for many protein structure which has strong MSA support, the reliability of the model for predicting the antibody-antigen complexes has not been sufficiently assessed. Here, we used 5 independent predictions from AlphaFold 3 to compare with a high-resolution cryo-EM structure of the dengue virus serotype 2 (DENV-2) E protein in complex with neutralizing antibody 2D22 (PDB: 8Y3H). AlphaFold 3 was able to reproduce the global fold of the E protein precisely; however, there was significant heterogeneity observed at the antibody-antigen interface. The predicted complexes were clustered into three separate binding modes, all three of which did not contain the experimentally determined epitope centered around residues A150 - 154 and A360 - 364. The predicted interfaces did not have the exact residue-level interactions that were seen in the cryo-EM structure. These results show that although AlphaFold 3 can reliably predict the backbone structure of antigens, it is still constrained in precisely positioning the location of the binding site of the antibody against flexible viral antigens. We show the importance of experimental proof and integrative modeling over the computational prediction in using them for guiding antiviral antibodies and vaccines design.
Introduction
Dengue fever is a major mosquito-borne viral disease, and it causes a substantial public health burden in tropical and subtropical areas around the world, with different outcomes of infection including self-limited febrile illness as well as severe haemorrhagic disease presenting with shock [1]. Although efforts to control dengue virus (DENV) are intensified, the virus still continues to spread in more than 100 countries, and the circulation is driven by various complicated host, virus and environment [1]. DENV is a enveloped flavivirus having a positive-sense RNA genome encapsidated within a 20 icosahedral protein capsid. The envelope (E) protein of the virus mediates the attachment of the virus to the host cell, receptor binding, and membrane fusion during viral entry, and it is the main target of neutralizing antibody responses [2, 3]. High-resolution experimental structures have revealed the domain structure of the E protein and its conformational changes during entry, which showed critical epitope surface that decided antibody binding specific and neutralize efficacy [3]. Such structural info on both counts—understanding how immune recognizes them and guiding rational vaccine and antiviral therapeutic development [4].
Potent neutralizing antibodies usually target conformational and quaternary epitopes on the E protein that are exposed on the surface of the mature virion; such interfaces typically straddle adjacent E subunits and are sensitive to the dynamic conformations of the protein [2]. Experimental structure determination by X-ray crystallography and cryo-EM has provided the information to define these interactions at the molecular level, and thus inform antibody-antigen recognition and immunogen design [3]. While homology-based and docking have made up for experimental studies, the predictability of these methods is hindered by the inherent flexibility of viral surface proteins and by their inability to represent complex intermolecular events [4].
Recent progress in deep learning-based structure prediction, most prominently in AlphaFold and its follow- ons, have significantly enhanced the accuracy of predicting protein structure from sequence [5, 6, 7]. The latest model architectures are, in principle, able to jointly predict the structures of protein complexes, including protein - protein interactions [7]. However, benchmarking studies have shown significant differences between predicted and experimental structures for antibody - antigen complexes, particularly at highly specific interface areas, which determine the conformation of immune recognition [8]. While deep learning models can frequently identify global fold topology with high confidence, they are less accurate when predicting paratope-epitope contacts and antigen structures [4, 9].
In this study we took the DENV-2 E protein, the serotype most often linked to severe disease to examine the performance of the AlphaFold structural predictions against high resolution cryo-EM reference structures [1]. By systematically comparing the predicted complexes with the experimentally determined ones, we were looking to find regions of structural differences and examine the consequences for antibody binding. Our analysis pointed out the main differences between the computational model and the empirical model, providing useful references to improve the prediction method and enhance the structural basis for future vaccine and therapeutic design.
Methods
Reference Structure and Input Sequences
A cryo-electron microscopy structure of the dengue virus serotype 2 envelope protein in complex with the Fab fragment of human antibody 2D22 Fibriansah, et al. [10] was used as a primary reference (PDB ID: 8Y3H). The amino acid sequences corresponding to the dengue viral proteins and human antibody proteins of the antigen-antibody complex were retrieved from the 8Y3H PDB file and used for subsequent structure prediction and analyses.
Structure Prediction with AlphaFold 3
Protein structure predictions were performed using the publicly accessible AlphaFold Server 3 (https:// alphafoldserver.com) (accessed on September 30, 2025). The viral envelope (E) protein, membrane polyprotein and the antibody heavy- and light-chain sequences were entered as separate polypeptide entities within a single AlphaFold 3 job. No additional experimental restraints, structural templates, or epitope information were supplied. The 5 top- ranked models from the AlphaFold 3 output were selected based on the server’s internal confidence metrics.
Structure Visualization and Analysis
The resulting predicted complexes were analyzed and evaluated through comparison with the experimentally validated antibody-antigen interface observed in the cryo- EM structure. Predicted complexes were structurally superposed onto the reference cryo-EM structure using UCSF ChimeraX with alignment performed on the E protein [11]. All subsequent structural comparisons, interface analyses, and figure preparation were conducted using PyMOL [12]. Results
Comparison of AlphaFold Prediction Structures
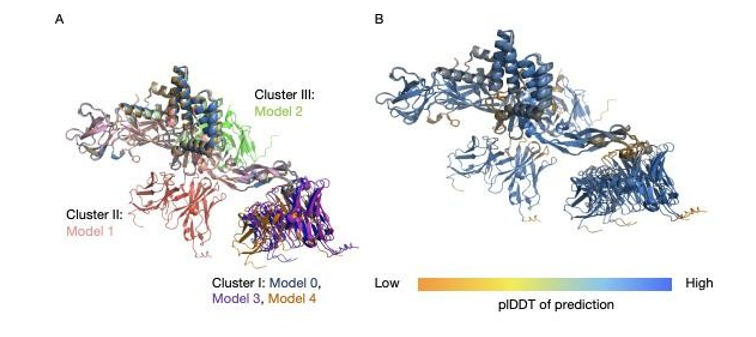
Structural superposition of five AlphaFold 3-predicted antibody-E protein complexes, aligned on the E protein, reveals a clear dissociation between antigen fold accuracy and antibody docking consistency (Figure A).Overall structure of E protein is highly conserved across all predictions, indicates robust and repeatable antigen model. Antibody has a very high degree of randomness in orientation and binding position, so the predicted interaction modes will be quite different from each other. This heterogeneity highlights the uncertainty in predicting the antibody - antigen interface despite the stable antigen backbone.
Among the five predictions, three models (Models 0, 3, and 4) make up the most common sampled binding configuration (Cluster I). In this cluster, the antibody faces a similar face of the E protein; heavy chain contacts are near residues E98 - 110; light chains are near E66 - 71. Although these models have a similar docking orientation, superposition shows some changes in the position of the antibody, indicating partial but not complete convergence on the same interface.
The other two predictions take on different binding modes that are not produced by other models. In Model 1 (Cluster II) the antibody is rotated away from the Cluster I binding face and binds residues near E121-125 via the heavy chain and a distal portion spanning E270-278 via the light chain. Model 2 (Cluster III) displays a third unique configuration, where the heavy-chain contacts center at E222-227, and the light-chain contacts near E130-135 on the side of the E protein. The absence of recurrence of these configurations indicates that the internal support is weak and that the breadth of interface variation captured by the model is extensive. Confidence mapping of the predicted complexes provides some more details on where the heterogeneity might be coming from (Figure 1B). For the most part, the antibody as well as E have high confidence scores per- residue, this is to be expected based on how well the accuracy is predicting a single proteins fold. However, there are localized regions of reduced confidence in both the antigen and the antibody, which contrast with the globally high confidence observed in the other parts of the structure. And importantly, those low confidence regions were en HT/or close to the ant Ub - Ag interfaces.
On the Eprotein there are decreases in confidence in surface exposed sections between 98 - 109, 146 - 159, 186 - 191, 271 - 281 and the flexible C terminal tail (492-495). Correspondingly, lower confidence is detected in antibody regions proximal to the predicted interfaces, including heavy- chain residues 54-58 and 101-105 and terminal segments of both chains. This spatial correspondence indicates that AlphaFold 3’s predictive uncertainty is concentrated at the interface, rather than in the global folds, highlighting a specific weakness in resolving antibody-antigen interaction regions.
Comparison Between AlphaFold Prediction Structure and PDB Results
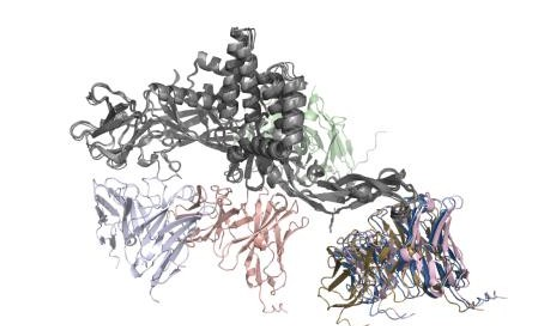
The AlphaFold 3-predicted antibody-E protein complexes were compared with the experimentally determined cryo- EM structure to evaluate the accuracy of predicted antibody- antigen binding sites (Figure 2). In the cryo-EM structure, antibody 2D22 engages two compact epitope regions on the E protein, centered at residues A150-154 and A360-364.
None of the predicted complexes place antibody contact regions near these experimentally validated sites. Instead, the predicted binding sites are distributed across multiple, topologically distinct regions of the E protein, corresponding to the heterogeneous docking configurations observed in the AlphaFold predictions. These predicted epitope regions are separated from the experimental binding site by entire domains of the E protein, indicating a lack of spatial overlap between predicted and true antigenic surfaces.
Comparison of Interface Interaction in PDB results and AlphaFold Prediction
To assess the accuracy of AlphaFold 3 in modeling antibody-antigen interactions, we compared the residue- level interaction chemistry of the predicted complexes with that of the experimentally resolved antibody-E protein structure. The Nature and Spatial Distribution of Interface Contacts, Emphasizing Hydrogen Bonding Patterns and the Localization of Antibody Paratopes Relative to the Antigen Surface (Figure 3).
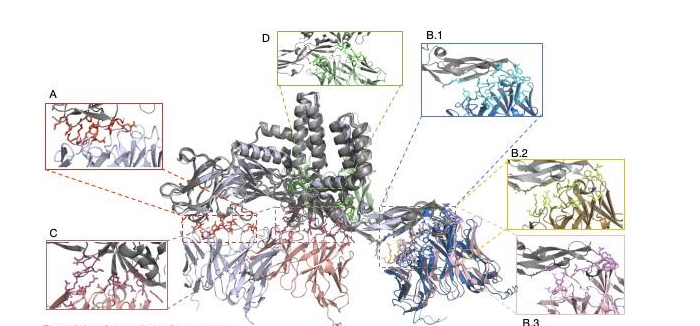
From the experimentally determined cryo-EM structure, a compact interface containing multiple intermolecular interactions is present (Figure 3A). There is no observed presence of disulfide bonds, covalent bonds or salt bridges. Instead, binding is via an interwoven hydrogen bonding network of the E protein between both antibody chains. E protein is bonded with heavy chain by A:VAL151 and G:ASN107 which are hydrogen bonded and light chain is bonded with epitope residues 150 - 154 and 360 - 364 via H:GLY30, H:ARG51, and H:ASN53 which are multiple hydrogen bonds. These interactions are perfectly centered on the heavy chain residues G101-107 and the light chain residues H30-33 and H51-54, and in combination with the experimentally validated epitope, the paratope achieves a geometrically complementary and energetically advantageous binding configuration.
In cluster I prediction (models 0, 3, 4), it looks like alphafold 3 is able to reproduce the general interaction mode observed in the experimental complex, interfaces seem to be mainly hydrogen bonding rather than covalent/electrostatic (Figure 3B.1 - 3B.3). The heavy chain is located close to the E-protein residues 98-110, and the light chain binds residues around 66-71, which are rich in polar side chains that can participate in hydrogen bonding. However, there is a kind of seeming agreement regarding the interaction chemistry, but this is at the cost of being incorrect as far as the spatial localization of the binding interface. The predicted hydrogen bond forms on the antigenic surface that cryo-EM structure does not have antibody-binding activity. As a result, the predicted interfaces do not coincide with the experimentally identified epitopes and cannot recapitulate the specific residue-level interaction network seen in Graph 3A, implying that the similarity in interaction types is coincidental chemical compatibility rather than correct epitope recognition.
Clusters II and III (Models 1 and 2) are even more inconsistent with the experimental interface and have their own unique and incompatible binding modes. In these models, the antibody contact sites have been redirected to alternate regions of the E protein at 121-125, 130-135, 222-227 and 270-278. Although these surfaces are capable of forming hydrogen bonds due to the polar and charged residues they contain, the resulting interfaces are broken and discontinuous, lacking the spatial order of the experimental complex. There is no consistent residue-level overlap with experimentally validated epitopes suggesting systematic misidentification of the antigenic binding sites among all of the predicted clusters.
Discussion
AlphaFold 3 is a big step forward for deep-learning- based structure prediction, it gets near-experimental accuracy for lots of proteins and complexes that follow strong evolutionary rules. In this study, AlphaFold 3 was able to reliably reproduce the global fold of the DENV-2 E protein and to consistently identify antibody paratope regions, thus confirming its power for modelling single protein structures and conserved interaction motifs. But, though these successes, AlphaFold 3 could not successfully locate the biologically meaningful antibody-antigen interface on the E protein-2D22 complex.
At the interface level analysis revealed that we have a clean dissociation for interaction chemistry being correct and spatial localization being correct. The predicted complexes all favored H-bonded interactions and excluded covalent bonds, disulfide bridges, and salt bridges as seen in the experiment. But still, all those chemically plausible interaction were systematically mapped to antigenic surfaces that do not take part in any kind of antibody binding according to the cryo-EM structure. And so the resulting interfaces were coincidentally chemically compatible rather than truly epitope recognizing. These limitations are in agreement with the known constraints of the MSA-driven deep learning models on applying to the antibody-antigen systems. Antibodies come about through somatic recombination and does not co- evolve with viral antigens, resulting in either poor or no evolutionarily coupled signals AlphaFold depends on to situate interacting surfaces. This problem is compounded by the conformational plasticity of the DENV E protein, and its neutralizing epitopes depend on the quaternary assembly and context of the virion. In the absence of any structural or biophysical constraint, AlphaFold samples energetically feasible yet biologically uninteresting docking conformations.
From a translational perspective, these findings have important implications for computational epitope mapping and structure-guided vaccine or antibody design. While AlphaFold predictions can provide valuable starting hypotheses, unrestrained models risk prioritizing non- neutralizing or cryptic surfaces, particularly for dynamic viral antigens such as flavivirus envelope proteins. Experimental validation thus remains indispensable for establishing biologically meaningful antibody-antigen interfaces.
Looking forward, further improvements will likely arise from integrative approaches that combine AlphaFold with experimental constraints, such as cryo-electron microscopy or cryo-electron tomography (cryo- ET), which capture native antigen organization on the virion surface. Incorporating density maps, mutational scanning, or antibody footprinting data could help guide interface localization and reduce biologically irrelevant docking solutions. While continued development of next-generation AlphaFold architectures is promising, careful benchmarking against high-resolution experimental structures will remain essential to ensure reliable application of deep learning models in antiviral antibody and vaccine development.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
A cryo-electron microscopy structure of the dengue virus serotype 2 envelope protein in complex with the Fab fragment of human antibody 2D22 Fibriansah, et al. [10] was used as a primary reference (PDB ID: 8Y3H). The amino acid sequences corresponding to the dengue viral proteins and human antibody proteins of the antigen-antibody complex were retrieved from the 8Y3H PDB file and used for subsequent structure prediction and analyses.
- An Efficient and Affordable Method for Isolating Bone Marrow- Derived Mesenchymal Stem Cells from Swiss Albino Mice
- Jugular-Applied Coherent Low-Level Laser Therapy Enhances Systemic Mitochondrial Metabolic Function and Antioxidant Response
- Role of OMC32 Polypeptide in Acrosin-Mediated Exocytosis during the Bovine Sperm Acrosome Reaction
- Association of Galectin-3 but not Laminin in Tamoxifen-Induced Growth Suppression in Breast Cancer MCF-7 Cells
- Effect of Different Wavelengths of Light on the Rate of Photosynthesis
- Nutritional, Therapeutic, and Environmental Effect of Oyster Mushrooms: An Editorial