Hemangioblastoma: Sporadic Cases and Von Hippel-Lindau's Disease: About 4 Cases and Literature Review
Hemangioblastomas (HGB) are rare tumors of the central nervous system (CNS) that originate from the vascular system. They represent 2 to 3% of brain tumors. The infratentorial (cerebellum, elongated marrow) or spinal (spinal marrow) are the locations of preferences (99%). These tumors account for 10% of all posterior cranial fossa brain tumors and are the most common primary posterior cranial fossa brain tumors in adults. Two-thirds of hemangioblastomas occur sporadically, onethird of cases in the context of familial neoplasia syndrome, von Hippel-Lindau disease (VHL). More than 80% of VHL patients will develop CNS hemangioblastoma during their lifetime and most of them (95%) will develop multiple hemangioblastomas. We report four cases of cerebelous hemangioblastoma treated in our neurosurgery department: two child brothers with VHL disease, from a mother treated for cerebelous hemangioblastoma, and two adults with sporadic hemangioblastoma. The 2 cases of hemangioblastomas in the context of VHL disease were, respectively, 13 years (Girl) and 22 years old (boy), from a mother with VHL disease and in both cases the first complaint was the symptoms of intra cranial hypertension syndrome associated with a cerebellar syndrome. Radiological exploration shows a cerebellar location for both cases associated with cervical spinal lesion in the case of the girl. Two sporadic male cerebellar hemangioblastomas cases aged 26 and 48, respectively. The 4 cases benefited from surgical exeresis with good evolution.
Introduction
Hemangioblastomas are tumors of the central nervous system. Considered grade I according to World Health Organization (WHO) [1, 2, 3, 4, 5, 6, 7]. They are benign vascular neoplasms that arise almost exclusively in the central nervous system but almost spare the supra tentorial region. They represent 1 to 2% of all intracranial tumors and show a predominant involvement of the cerebellum [8, 9]. Hemangioblastomas may be present either in the context of von Hippel-Lindau disease (VHL), or more often as a sporadic lesion without a family history [10, 11]. Von Hippel-Lindau disease (VHL) is an inherited disease caused by genetic mutations of the VHL gene on chromosome 3 (WG 2002). The mutated VHL gene is transmitted from generation to generation in a dominant autosomal way with almost complete penetration [12]. The incidence of VHL ranges from 1 in 36,000 to 1 in 45,000 live births. Generally, VHL occurs for the first time in the second decade of life. At the time of detection, 50% of patients are symptomatic [13]. Cerebellar hemangioblastoma (35%) are the most common manifestation of VHL disease [14, 15, 16].
These tumors with their benign histological character tend to compress the adjacent parenchyma rather than infiltrating it [17, 18]. Surgical excision is a preferable, safe and timely curative strategy for preventing neurologic decline [19].
Observations
Observation No. 1 (A. M)
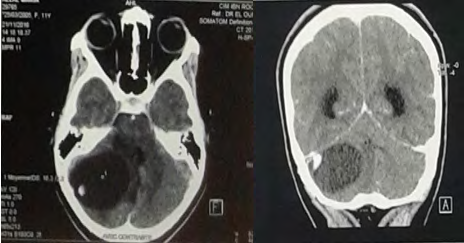
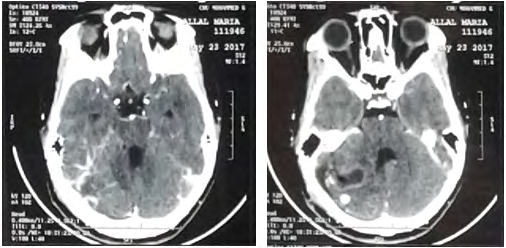
A.M, 13, the youngest of a sibling of 4, from a non- consanguineous marriage, with a history of maternal death from a brain tumor and a brother diagnosed with hemangioblastoma. Clinically the patient was admitted for a decompensated intracranial hypertension syndrome affecting the vision with cerebellar syndrome, evolving 2 weeks before her admission. The examination finds a decrease in visual acuity in bilateral, convergent strabismus, left nystagmus, instable balance, without sensory-motor deficit. The funduscopic examination, showed papillary edema. Cerebral CT without injection (Figure 1) showed a tumor process of the right cystic cerebellar Hemisphere with two fleshy portions compressing 4V and responsible for triventricular hydrocephalus.
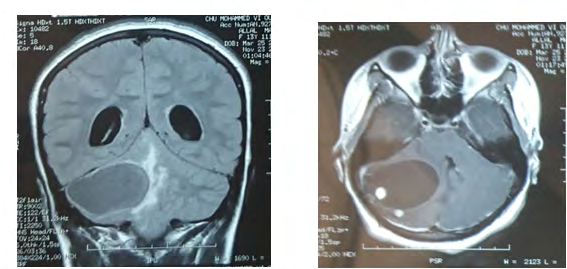
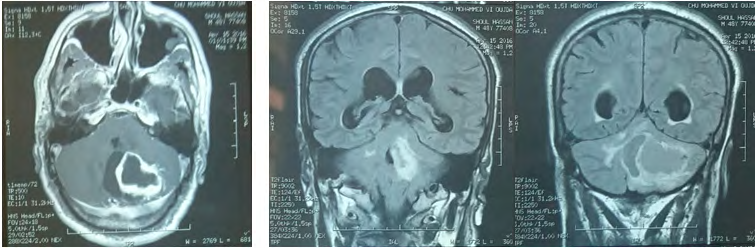
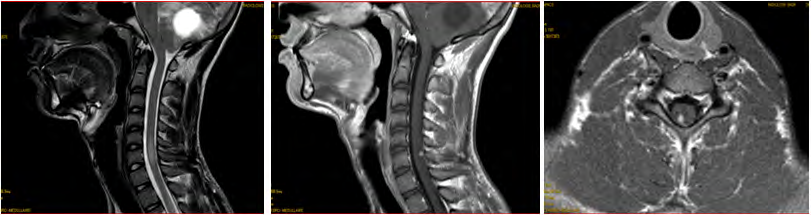
Brain MRI (Figure 2) shows a 49 mm of diameter cerebellar mass of highly hypo-intense cystic T1, hyper-intense T2 with two parts of 1 cm diameter iso-intense in signality in T1, hypersignal T2 highly enhanced by the contrast product, responsible for a triventricular hydrocephalus. Medullary MRI (Figure 3) objectifying a medullary nodule next to L3- L4 described in hyposignal T2 and strongly enhanced after injection of contract product. The patient was operated on by an initial lateralized occipital on the right which allowed an evacuation of the cyst and subtotal exeresis of the two fleshy nodules. Immediate surgical consequences were simple. The evolution was marked by the reappearance of the same clinical picture made of HTIC syndrome after a remission of 6 months. Control eye background: showed papillary edema.
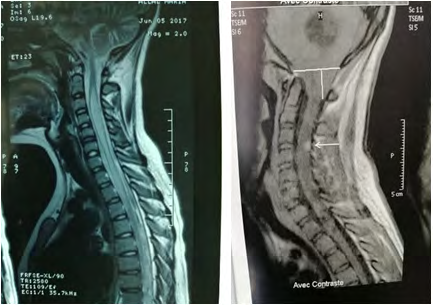
MRI control (Figure 4) showed recurrence of the tumor process in the same solidocystic form of 54.8 mm of large axis. She was re-operated the same way first with a macroscopically total exeresis (Figure 5), at post-op J-3: the patient presented complicated back pain of paraparesis allowing walking with a medullary claudication.
Two years later, the patient is seen in complete clinical remission.
- Histopathology: Hemangioblastoma.
- Genetic study: confirms the presence of VHL mutation.
Observation No 2 (F.M)
F.M, 26 years old without a history of consanguineous marriage, epileptic (without an etiological diagnosis of epilepsy) since the age of 16 with an antiepileptic treatment. The patient admitted for an intracranial Hypertension Syndrome associated stato-kinetic cerebellar syndrome evolving 10 days before admission. The funduscopic examination: normal. Cerebral CT without injection showed an expansive process of the right cerebellar hemisphere measuring 42x47x37 mm with mass effect on the 4th ventricle responsible for tri-ventricular hydrocephalus. Brain MRI (Figure 6), shows right hemispherical cerebellar lesion, predominantly cystic hypointense T1, hyper-intense T2 with a solid component of 14x5 mm major diameter isosignal T1 hypersignal T2 strongly enhanced after contrast, responsible for a tri-ventricular hydrocephalus.
The patient was operated on first by right suboccipital lateral who allowed an evacuation with macroscopically total excision of the cerebellar process. The patient was put on IPP, corticosteroid, paracetamol Phenobarbital carbamazepine and sodium valproate.
The postoperative course was simple. • Pathology: hemangioblastoma.
The evolution was marked by the regression of the symptomatology.
Observation No 3 (S.H)
S .H, 48 years old, known as diabetic type II for 12 years. The patient was admitted for an intracranial Hypertension Syndrome with a stato-kinetic cerebellar syndrome and vertigo evolving for 4 weeks before his admission. • The funduscopic examination: minimal diabetic retinopathy without papillary edema.
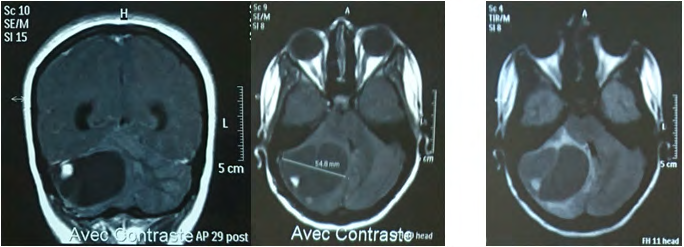
Biological assessment found a polycythemia with a hematocrit at 61.2% and hemoglobin at 20g / dl. Abdominal ultrasound showed a steatosis liver with 2 left renal lithiasis with small dilation of the pyelocaliceal cavities. Cerebral CT scan (Figure 7), without injection of contrast, showed an expansive process of the left cerebellar hemisphere measuring 40x29 mm, enhanced wall after injections of contrast, achieving a mass effect on the 4th ventricle and responsible for a tri-ventricular hydrocephalus. They associate with pre-lesional edema in glove. Cerebral MRI (Figure 8) found a left cerebellar solido-cystic tumor process left: cyst portion hypointense in T1 with enhanced wall after injections of contrast. with peri-lesional edema described in T2 hypersignal.
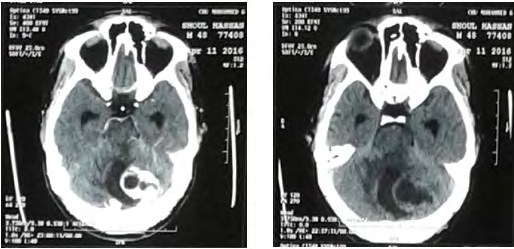
The patient was operated by a left lateral sub-occipital approach which allowed an evacuation with macroscopically total excision of a cerebellar process. The suites were simple except a stay in intensive care for 3 days for arterial tension control.
The patient was put on the regimen of insulin therapy, corticosteroids. • Histopathology: hemangioblastoma The evolution was marked by the regression of HTIC syndrome and cerebellar syndrome.
Observation N 4 (A.A)
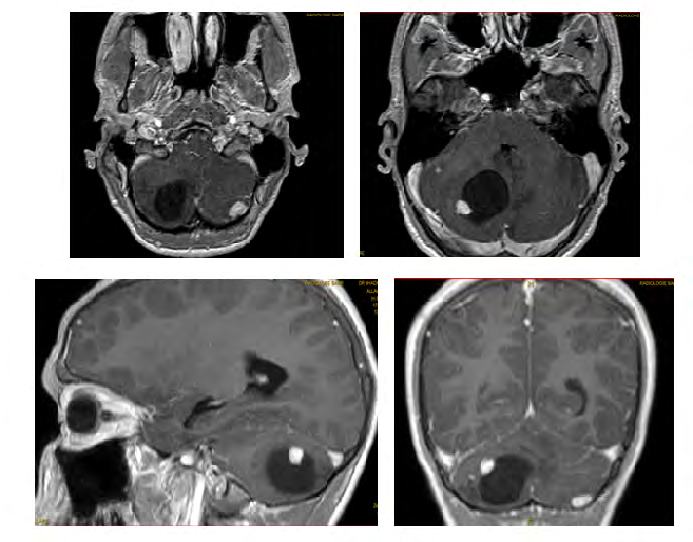
A.A, 20 years old, from a non-consanguineous marriage with an antecedent of a maternal death from a brain tumor and a sister followed for a hemangioblastoma. The patient had an intracranial hypertensive syndrome that had been evolving for 1 month and a half before admission. The funduscopic examination without particularity. Cerebral CT showed a solidocystic process in the right cerebellar hemisphere and a second nodular process in the left side. Cerebro-medullary MRI (Figures 9 & 10) found two solidocystic processes in the right cerebellar hemisphere and a second left-sided nodular process, with two extensive medullary lesions of C5-C6 enhanced after contrast injection.
The patient was operated on by the medial suboccipital approach with macroscopically total excision of both processes. The suites were simple.
- Histopathology: hemangioblastoma.
- Genetic study: confirms the presence of VHL mutation.
Discussion
Hemangioblastomas are the most common tumors associated with VHL disease. In more than 80% of patients with the disease can develop craniospinal hemangioblastoma during their lifetime (with an average age of 33 years) [20] and more than 90% of patients with hemangioblastoma develop multiple CNS hemangioblastomas [21]. Craniospinal hemangioblastomas are almost entirely found (99%) in the cerebellum, brainstem and spinal cord. Hemangioblastomas occur most often in the cerebellum (45% to 50%) spinal cord (40% to 45%) cervical, and less frequently in the brainstem (5% to 10%) (LI. 2002) [22]. About 66% of CNS hemangioblastomas develop sporadically and the rest, in the context of VHL disease. The signs or symptoms of their clinical manifestations are associated with the anatomical location of the tumor. No reliable threshold for the size of the tumor can predict the onset of symptoms and the need for treatment [23, 24]. In the case of cerebellar disease, the patient may experience headache, dizziness, walking and speech problems, nystagmus, diplopia, ptosis, dysmetria, paralysis of the ninth cranial pair and paroxysmal hypertension. Erythrocytosis is rarely reported and is secondary to a constituent activation of erythropoietin caused by impaired degradation of the hypoxia-induced factor (HIF) [25]. In the case of spinal cord injury, symptoms may include neurogenic pain, multiple sensory deficits, proprioceptive disorders, paraparesis, paresthesia, muscle atrophy and medullary hypertonia.
Contrairement aux patients VHL ou aux membres de leur famille qui doivent être évalués à la recherche d’hémangioblastomes sur la totalité du névraxe, les cas sporadiques sont généralement diagnostiqués presque toujours tardivement au moment de la survenue des symptômes. La date d’apparition des symptômes avant le diagnostic variait d’un demi-mois à 12 mois. Unlike VHL patients or family members who need to be evaluated for hemangioblastomas on the entire neural-axis, sporadic cases are usually diagnosed almost always late at the time of the occurrence of Symptoms. The date of onset of symptoms prior to diagnosis ranged from half a month to 12 months [26].
Differences in the molecular and genetic origins of hemangioblastomas may explain why patients with sporadic lesions have much later results than those with VHL-related tumors [27]. Genetic testing is performed when VHL disease is suspected due to clinical presentation in a patient or family member. After conducting a medical and family history investigation, a clinical examination should be performed prior to the mutation test. Peripheral blood samples are obtained for the mutation test. To identify cases of mosaism, it is preferable to remove peripheral blood from the affected parent [28]. When mutation screening does not identify a mutation in peripheral blood, DNA testing from other tissues and/or VHL manifestations may be necessary to confirm the diagnosis.
Previously, hemangioblastomas were classified as unknown histogenesis tumors. But recent studies indicate that these tumors originate from stromal cells derive from angiomatous angimamamatous precursors that block their maturation in patients with VHL disease [29, 30]. Similar to embryologically normal hemangioblasts, stromal hemangioblastoma cells express brachyure, VEGF 2 (Flk-1) and stem cell leukemia. Tumor-derived stromal cells (hemangioblasts) can differentiate into hematopoietic and endothelial progenitors [31]. MRI is the exam of choice to explore the posterior cranial fossa. In case of hemangioblastomas, it helps to better clarify their cystic and solid components. The injection of gadolinium can reveal nodules that are not or are not visible on CT scan (JL. 2006). Thus, the cyst appears in the form of a very limited formation in homogeneous hyposignal T1, hypersignal T2 and hyposignal Flair. For the nodular part appears in intermediate signal in T1 and rises strongly after contrast. The wall nodule is usually sits at the cyst on contact with a leptomeninges surface. There are solid forms with predominance of the fleshy part (JL. 2006).
Occasionally, a subtraction angiography can be used to assess the vascularization of the tumor, especially in larger lesions. On angiography, in the presence of hemangioblastomas show an enlarged nourishing artery, early drainage of the veins and an enhancement of the tumor that persists in the venous phase. Given the strong association between hemangioblastomas and VHL, patients with newly diagnosed hemangioblastoma should be screened for VHL. Once a diagnosis of CNS hemangioblastoma has been made, brain-spinal imaging is required for further location. In search of systemic lesions associated with VHL, abdominal imaging and a cross-eye background should be performed as part of the initial screening [32]. In patients with a single hemangioblastoma of the central nervous system, no family history of VHL disease or any signs of systemic manifestations of VHL, only 4% of patients will have a detectable VHL mutation. These patients are usually under 40 years of age and a search for the VHL mutation is recommended.
In addition to abdominal imaging, 24-hour catecholamines in the urine and an eye exam. Long-term monitoring of patients with CNS hemangioblastoma depends largely on age and genetic balance. Patients with a germ line mutation of VHL should be monitored for life, in addition to screening at-risk relatives [33]. Given the link between young age at the time of presentation and VHL disease, younger patients should be closely monitored, even for negative mutation analysis, as genetic mosaicism can lead to false negative testing. In addition, the risk of FAMILY VHL-related disease in a patient with solitary hemangioblastoma over 50 years of age is quite low. Complete resection is the treatment of choice for symptomatic hemangioblastoma. Because of the high intraoperative and postoperative hemorrhagic risk, and the curative nature of a complete resection [34].
Preoperative embolization to reduce intraoperative bleeding is not commonly used and may be associated with an additional risk. Stereotactic or craniospinal radiotherapy has been used in patients who are not candidates for surgery. However, long-term follow-up indicates that radiation-borne hemangioblastomas will continue to grow in a proportion similar to that expected by natural history [35, 36].
Conclusion
Despite their benign nature, hemangioblastomas are the first etiology of morbidity in patients with VHL due to the anatomical location of these tumors and/or mass effect [37, 38, 39]. Recent studies have led to a better definition of natural history, underlying biological characteristics and long-term outcomes after surgical resection of these lesions, resulting in an evolution of treatment strategy and redefinition of surgical indications. This has led to a considerable improvement in the quality of life of patients.
References
-
Cornelius JF, Saint-Maurice JP, Bresson D, George B, Houdart E (2007) Hemorrhage after particle embolization of hemangioblastomas: comparison of outcomes in spinal and cerebellar lesions. J Neurosurg 106(6): 994-998.
-
Tampieri D, Leblanc R, TerBrugge K (1993) Hemangioblastomas: Preoperative embolization of brain and spinal. Neurosurgery 33(3): 502-505.
-
Wanebo JE, Lonser RR, Glenn GM, Oldfield EH (2003) The natural history of hemangioblastomas of the central nervous system in patients with von Hippel-Lindau disease. J Neurosurg 98(1): 82-94.
-
Ganguly R, Dornbos D, Finlay JL, Lonser RR (2019) Hemangioblastoma and von Hippel-Lindau Disease. In: Tonn JC, Reardon D, Rutka J, Westphal M (Eds.) Oncology of CNS Tumors. Springer, Cham, pp: 321-329.
-
Sung DI, Chang CH, Harisiadis L (1982) Cerebellar hemangioblastomas. Cancer 49(3): 553-555.
-
Maher ER, Yates JRW, Harries R, Benjamin C, Harris R, et al. (1990) Clinical features and natural history of von Hippel-Lindau disease. Q J Med 77(2): 1151-1163.
-
Neumann HP, Eggert HR, Weigel K, Friedburg H, Wiestler OD, et al. (1989) Hemangioblastomas of the central nervous system. A 10-year study with special reference to von Hippel-Lindau syndrome. J Neurosurg 70(1): 24- 30.
-
Conway JE, Chou D, Clatterbuck RE, Brem H, Long DM, et al. (2001) Hemangioblastomas of the central nervous system in von Hippel-Lindau syndrome and sporadic disease. Neurosurgery 48(1): 55-63.
-
Niemela M, Lemeta S, Summanen P, Bohling T, Sainio M, et al. (1999) Longterm prognosis of haemangioblastoma of the CNS: impact of von Hippel–Lindau disease. Acta Neurochir (Wien) 141(11): 1147-1156.
-
Chen-Chieh Liao, Yu-Hua Huang (2014) Clinical features and surgical outcomes of sporadic cerebellar hemangioblastomas. Clinical Neurology and Neurosurgery 125: 160-165.
-
Choyke PL, Glenn GM, Walther MM, Patronas NJ, Linehan WM, et al. (1995) Von Hippel-Lindau disease: genetic, clinical, and imaging features. Radiology 194(3): 629- 642.
-
Maher ER, Iselius L, Yates JR, Littler M, Benjamin C, et al. (1991) Von Hippel-Lindau disease: a genetic study. J Med Genet 28(7): 443-447.
-
Poulsen ML, Budtz-Jørgensen E, Bisgaard ML (2010) Surveillance in von Hippel-Lindau disease (vHL). Clin Genet 77(1): 49-59.
-
Neumann HP, Wiestler OD (1991) Clustering of features of Von Hippel-Lindau syndrome: evidence for a complex genetic locus. Lancet 337(8749): 1052-1054.
-
Maddock IR, Moran A, Maher ER, et al. (1996) A genetic register for von Hippel-Lindau disease. J Med Genet 33(2): 120-127.
-
Evans DG, Howard E, Giblin C, Clancy T, Spencer H, et al. (2010) Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A 152A(2): 327-332.
-
Karabagli H, Genc A, Karabagli P, Abacioglu U, Seker A, et al. (2010) Outcomes of gamma knife treatment for solid intracranial hemangioblastomas. J Clin Neurosci 17(6): 706-710.
-
Sayer FT, Nguyen J, Starke RM, Yen CP, Sheehan JP (2011) Gamma knife radiosurgery for intracranial hemangioblastomas-outcome at 3 years. World Neurosurg 75(1): 99-105.
-
Lonser RR, Weil RJ, Wanebo JE, Devroom HL, Oldfield EH (2003) Surgical management of spinal cord hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg 98(1): 106-116.
-
Feletti A, Anglani M, Scarpa B, et al (2016) Von Hippel- Lindau disease: an evaluation of natural history and functional disability. Neuro Oncol 18(7): 1011-1020.
-
Feldman MJ, Sizdahkhani S, Edwards NA, Merrill MJ, Chaudhury AR, et al. (2016) Loss of quiescence in von Hippel-Lindau hemangioblastomas is associated with erythropoietin signaling. Sci Rep 6: 35486.
-
Murota T, Symon L (1989) Surgical management of Hemangioblastoma of the spinal cord: a report of 18 cases. Neurosurgery 25(5): 699-707.
-
Lonser RR, Butman JA, Huntoon K, Asthagiri AR, Wu T, et al. (2014) Prospective natural history study of central nervous system hemangioblastomas in Von Hippel- Lindau disease. J Neurosurg 120(5): 1055-1062.
-
Crespigio J, Berbel LCL, Dias MA, Berbel RF, Pereira SS, et al. (2018) Von Hippel-Lindau disease: a single gene, several hereditary tumors. J Endocrinol Invest 41: 21-31.
-
Wiesener MS, Seyfarth M, Warnecke C, Jurgensen JS, Rosen berger C, et al. (2002) Paraneoplastic erythrocytosis associated with an inactivating point mutation of the Von Hippel-Lindau gene in a renal cell carcinoma. Blood 99(10): 3562-3565.
-
Sankaredja J, Sankaredja J, Brac B, Thines L, Baroncini M, et al. (2014) Epidemiology, treatment and follow-up of central nervous system hemangioblastomas in von Hippel-Lindau disease. Rev Neurol 170(4): 288-296.
-
Binderup MLM, Bisgaard ML, Harbud V, et al. (2013) Von Hippel–Lindau disease (vHL). National clinical guideline for diagnosis and surveillance in Denmark. Dan Med J 3rd edition. Dan Med 60(12): B4763.
-
Prashant Chittiboina, Russell R Lonser (2015) Von Hippel-Lindau disease. Handbook of Clinical Neurology 132: 139-156.
-
Park DM, Zhuang Z, Chen L, Szerlip N, Maric I, et al. (2007) von Hippel-Lindau Disease-Associated hemangioblastomas are derived from embryologic multipotent cells. PLoS Med 4(2): 60.
-
Richard S, Parker F, Aghakhani N, Allegre G, Portier F, et al. (2005) Von Hippel-Lindau disease: recent advances in genetics and clinical management. J Neuroradiol 32(3): 157-167.
-
Ding X, Zhang C, Frerich JM, Germanwala A, Yang C, et al. (2014) De novo VHL germline mutation detected in a patient with mild clinical phenotype of von Hippel- Lindau disease. J Neurosurg 121(2): 384-386.
-
Singh AD, Nouri M, Shields CL, Shields JA, Smith AF (2001) Retinal capillary hemangioma: a comparison of sporadic cases and cases associated with von Hippel- Lindau disease. Ophthalmology 108(10): 1907-1911.
-
Woodward ER, Wall K, Forsyth J, Macdonald F, Maher ER (2007) VHL mutation analysis in patients with isolated central nervous system haemangioblastoma. Brain 130(Pt 3): 836-842.
-
Wind JJ, Bakhtian KD, Sweet JA, Mehta GU, Thawani JP, et al. (2011) Long-term outcome after resection of brainstem hemangioblastomas in von Hippel-Lindau disease. J Neurosurg 114(5): 1312-1318.
-
Asthagiri AR, Mehta GU, Zach L, Li X, Butman JA, et al. (2010) Prospective evaluation of radiosurgery for hemangioblastomas in von Hippel–Lindau disease. Neuro Oncol 12(1): 80-86.
-
Chang SD, Meisel JA, Hancock SL, Martin DP, McManus M, et al. (1998) Treatment of hemangioblastomas in von Hippel-Lindau disease with linear accelerator-based radiosurgery. Neurosurgery 43(1): 28-34.
-
Sarrazin JL (2006) Tumeurs de la fosse poste´rieure. J Radiol 87(6): 748-763.
-
Malis LI (2002) Atraumatic bloodless removal of intramedullary hemangioblastomas of the spinal cord. J Neurosurg 97(1): 1-6.
-
Kaelin WG (2002) Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer 2(9): 673-682.
- Management of Chronic Insertional Achilles Tendinopathy Using Flexor Hallucis Longus Tendon Transfer in Patients Over 50 Years of Age: A Four-Case Series Following the CARE Guidelines
- Application of Induced Pluripotent Stem Cells in Bone Tissue Engineering: Current Status and Prospects
- Surgical Management of Upper Thoracic Esophageal Squamous Cell Carcinoma with Concomitant Hypersplenism: Integration of Chai's Supra-Thoracic Apex Technique with Laparoscopic Splenectomy - A Technical Innovation Case Study with Systematic Review
- Evaluation of Masticatory Functional Efficiency of Stomatognathic System in Patients Undergoing Open Reduction Internal Fixation for Treatment of Pan-Facial Trauma: A Prospective Study
- Hepatic Abscess Secondary to Appendiceal Phlegmon an Unusual Complication of Appendiceal Phlegmon
- Report of Lumboperitoneal (LP) Shunt Procedure in Over Decades Experiences, Systematic Narrative Review