Intractable Epilepsy and Gene Study
Objectives: Approximately 20% of epileptic children resist anti-seizure medications (ASMs). Management of these cases is complex and needs many pre-requisites. A Gene study is a part of the assessment in epilepsy cases and adjusts according to the physician’s decision. We conducted a gene study and assessed the response to ASMs. Material & Methods: All the ASMs resistant cases that were suspected of genetic epilepsy, did not have metabolic/structural etiology or neurodegenerative disease, and were referred for genetic study between Jan 2014 to Dec 2021 were enrolled. The records of 52 cases were assigned in our registry. The name of the gene extracted and the response to ASMs evaluated. The mean age was 6.65 years old; 24 (46.2%) were boys and 28 (53.8%) were girls. Results: The most common found genes were SCN1A, CAD1, IQSEC2, SLc6A1, AP3B2, SLC25A22, and KCNJ10. The complete response rate was seen in 20 cases (38.46%). Conclusion: In conclusion, further studies should be profit to make a link between gene type and drug response and achieve a better seizure- free response. If this happened, precision medicine would be more achievable
Introduction
Drug- resistant epilepsy is defined by the international league against epilepsy (ILAE) as the failure of adequate Conceptual Paper trials of two tolerated, appropriately chosen and used anti- seizure medications ASMs [1]. Approximately 7 to 20 percent of children with epilepsy are resistant to ASMs [2]. These cases also have a higher rate of morbidity and mortality, seven to twenty-fold higher than the average children [3, 4]. Many risk factors report for drug- resistant epilepsies, including developmental delay at diagnosis, abnormal brain imaging, and age of onset [5]. Beside these, there is a link between genetic variations and drug- resistant seizures [6].
The genetic differences affect the tolerability, efficacy, safety, and duration of action of a drug. However, the role of drug prescription according to the genetic variation in these patients is weakly investigated.
Material and Methods
This study was conducted in Ghaem hospital pediatric neurology center, using the handwritten hospital registry of this medical center. All the ASMs resistant cases that were suspected of genetic epilepsy, did not have metabolic/ structural causes or a neurodegenerative disease, and were referred for genetic study between Jan 2014 to Dec 2021 enrolled. With this regard, a sample of 52 confirmed cases was gathered.
All the medical records were reviewed for further information. Data including age, gender, family history, and medical history extracted. Moreover, the age of epilepsy onset, treatment onset, the number of seizures, frequency, and type of seizures were recorded. The type of mutated gene was assigned, and the response rate to medical therapy classified into complete response (seizure- free), excellent (reduction in seizure episodes>75%), fair (reduction in seizure episodes of 50-75%), weak (reduction in seizure episodes of 25-50%), and no response (reduction in seizure episodes <25%). All the records of the patients were anonymized and coded in order to be secret. All the steps of the study were following Helsinki’s declaration and approved by the ethics committee of Mashhad University of Medical Sciences (Code: IR.MUMS. MEDICAL.REC.1400.506).All the extracted data was entered in SPSS software version 20. The frequency and percent of the qualitative data and the mean and standard deviation of the quantitative data calculated. The data is demonstrated in the form of plots and tables.
Results
52 patients with a mean age of 6.65 years old enrolled in this study. Among these cases, 24 (46.2%) were boys, and 28 (53.8%) were girls. In the case of family history, 22 cases (42.3%) had a similar positive family history, and only in 15 cases (28.8%), their parents had no consanguinity. Table 1 demonstrates demographic family history, mental and physical development, and other accompanying underlying problems.
| Feature | Statistics | |
|---|---|---|
| Age (years; mean±SD) | 6.65±4.09 | |
| Gender N (%) | Male | 24 (46.2) |
| Gender N (%) | Female | 28 (53.8) |
| Development status (age<6 years) | Delayed | 50 (96.2) |
| Development status (age<6 years) | Natural | 2 (3.8) |
| School attendance N (%) | Normal school | 2 (3.9) |
| School attendance N (%) | Special school | 24 (46.1) |
| School attendance N (%) | Not attending school | 26 (50.0) |
| Physical status N (%) | Independent | 3 (4.3) |
| Physical status N (%) | Dependent to other | 49 (95.7) |
| Intellectual status N (%) | Delayed | 50 (96.2) |
| Intellectual status N (%) | Natural | 2 (3.8) |
Table 1: Demographic, mental and physical development.
Table 2 also shows data regarding the age of seizure onset, type of seizure, and frequency. The frequency of seizure was daily in 27 (51.9%) cases, and the most frequent type was epileptic spasm (39 patients (56.4%).
| Feature | Statistics | |
|---|---|---|
| Age of seizure onset (months; mean±SD) | 14.48±21.11 | |
| Age of starting medication (months; mean±SD) | 16.19±22.88 | |
| Number of different types of seizure (mean±SD) | 1.88±0.83 | |
| Seizure frequency N (%) | Daily | 27 (51.9) |
| Weekly | 10 (19.2) | |
| Monthly | 7 (13.5) | |
| Unspecified | 6 (11.5) | |
| Other | 2 (3.8) | |
| Type of seizure N (%) | Spasm | 39 (56.4) |
| General tonic | 24 (34.8) | |
| +General colonic | 17 (26.0) | |
| Focal tonic | 18 (24.6) | |
| Focal myoclonic | 13 (18.8) | |
| Focal with awareness | 6 (8.6) | |
| Absence | 5 (7.2) | |
| Unclassified | 1 (1.4) | |
| Focal without awareness | 1 (1.4) | |
| Focal colonic | 1 (1.4) | |
| Atonic | 1 (1.4) |
Table 2: Age of seizure onset, Type of seizures and frequency.
The mean number of prescribed drugs was 2.56±1.26. The most frequent drug was phenobarbital, prescribed in 40 cases (78.9%). Table 3 shows the details of the treatment in these patients.
Table 3 demonstrates detected genes in the studied population. The most frequent gene was SCN1A (four cases).
| Identified genes | Frequency | Percent |
|---|---|---|
| KeTD7 | 1 | 1.9 |
| CAD1 | 2 | 3.8 |
| KIF7 | 1 | 1.9 |
| FRRS1L | 1 | 1.9 |
| KeNT8 | 1 | 1.9 |
| SCN1A | 4 | 7.7 |
| OCLN | 1 | 1.9 |
| WWOX | 1 | 1.9 |
| TBC1D24 | 1 | 1.9 |
| DEE94 | 1 | 1.9 |
| IQSEC2 | 2 | 3.8 |
| SLc6A1 | 1 | 1.9 |
| TBe1D24 | 1 | 1.9 |
| PCDH19 | 1 | 1.9 |
| AP3B2 | 2 | 3.8 |
| SLC2SA22 | 2 | 3.8 |
| ScN2A | 1 | 1.9 |
| SCN8A | 1 | 1.9 |
| STXBP1 | 1 | 1.9 |
| DEE28 | 1 | 1.9 |
| CDKI5 | 1 | 1.9 |
| CXorf56 | 1 | 1.9 |
| ST3GAL3 | 1 | 1.9 |
| CACNA1A | 1 | 1.9 |
| NPC1hom | 1 | 1.9 |
| NPC1 | 1 | 1.9 |
| TPP1 | 1 | 1.9 |
| ARD1B | 1 | 1.9 |
| SPTBN2 | 1 | 1.9 |
| KCNJ10 | 2 | 3.8 |
| CLN5 | 1 | 1.9 |
| PDE10A | 1 | 1.9 |
| ARSA | 1 | 1.9 |
| DPM3 | 1 | 1.9 |
| NGLY1 | 1 | 1.9 |
| AMPD2 | 1 | 1.9 |
| MPC1 | 1 | 1.9 |
| CRIPT | 1 | 1.9 |
| MRPS35 | 1 | 1.9 |
| Prose | 1 | 1.9 |
| NALCN | 1 | 1.9 |
| AP4M1 | 1 | 1.9 |
| PIGL p.Met244Leu | 1 | 1.9 |
| SYNJ1 | 1 | 1.9 |
Table 3: Detected genes in the studied population.
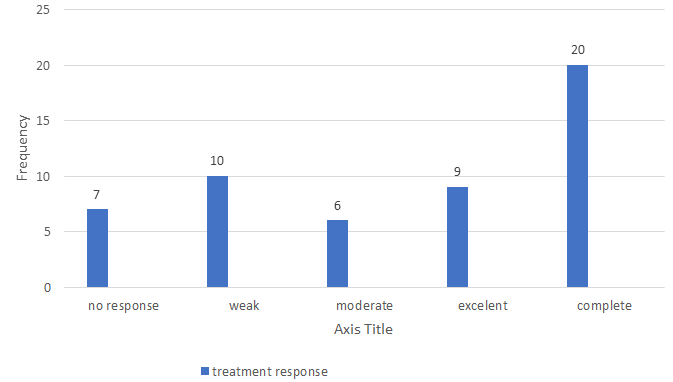
Figure 1 shows the frequency of different response rates to anti-epileptic drugs. 38.46% of the cases had a complete response.
Electroencephalography findings were normal in three cases (11.2%) and abnormal in 24 cases (88.8%). Moreover, in case of magnetic resonance imaging was normal in 24 patients (55.8%) 16 cases (37.2%) showed atrophy, two cases (4.6%) had hemi-megalocephaly, and one (2.3%) had cerebellar hypoplasia.
Table 4 shows different genes and different responses. None of the cases with a mutation in SCN1A or SCN2A received carbamazepine or phenytoin as medication. Moreover, 42 patients intended to have another child, and all of them provided genetic assessments in this regard.
| Complete | Excellent | Moderate | Weak | No response | |
|---|---|---|---|---|---|
| CAD1 | 1 (50.0) | 0 (0.0) | 0 (0.0) | 1 (50.0) | 0 (0.0) |
| SCN1A | 2 (50.0) | 0 (0.0) | 0 (0.0) | 2 (50.0) | 0 (0.0) |
| IQSEC2 | 0 (0.0) | 2 (100.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| SLc6A1 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (100.0) | 0 (0.0) |
| AP3B2 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (50.0) | 1 (50.0) |
| SLC25A22 | 2 (100.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| KCNJ10 | 1 (50.0) | 1 (50.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Table 4: Common genes and response rate.
Discussion
We found different genes with different responses to drug treatment. The most frequently found gene mutation in our study was in SCN1A, with a frequency of four cases. This gene encodes alpha subunit of voltage-gated sodium channels. The literature also proposes this gene as one of the most mutated genes in drug- resistant epilepsy and has been reported to have the responsibility in many disorders, including Generalized Epilepsy with Febrile Seizure Plus (GEFS+), Dravet Syndrome (DS), Doose syndrome (MAE), Borderline Severe Myoclonic Epilepsy of Infancy (SMEB), infantile spasms, and some other infantile epileptic disorders [7, 8, 9]. It is reported that patients with SCN1A and SCN2A mutations show no response to carbamazepine and phenytoin, as these drugs also act on sodium channels [10]. Two out of four of the studied cases become seizure- free in our study, but two showed a weak response. Usluer S, et al. [11] also studied 46 epilepsy cases with SCN1A gene mutation and reported that 26 patients (56.52%) had no response to the ASMs. It can hypothesize that factors other than not receiving carbamazepine should consider in management of patients with SCN1A mutation. Even different polymorphism of this gene may exhibit different properties. Another study suggested that SCN1A IVS5-91 rs3812718 G>A polymorphism is associated with even responsiveness to ASMs monotherapy [12].
SLC25A22 found in two cases in our study, which is in the mitochondrial transporter family. This gene reportedly responsible for early infantile epileptic encephalopathy, especially Migrating Partial Seizures in Infancy. There is a case report with SLC25A22 mutation and resistance to phenobarbital, carbamazepine, vigabatrin, pyridoxal five n -phosphate, and levetiracetam; however, an exact link with this regard is absent [13]. The mutation in this gene reported in many disorders, including Ohtahara syndrome, early myoclonic epilepsy, and epilepsy of infancy with migrating focal seizures. It is reported that in all disorders, a ketogenic diet may play a crucial role, and this can be more important than drug change [14]. We had two excellent response cases; however, due to the retrospective manner of the study, the efficacy of the ketogenic diet cannot be detected.
The mutation KCNJ10 gene was another found mutation in two cases of our population. KCNJ10 is located on chromosome 1q22–23 and encodes inward rectifier potassium ion channel protein (Kir4.1). This protein is responsible for potassium influx into glial cells from intracellular space, in order to maintain a resting state. Besides the fundamental role in developing childhood epilepsy, some genetic variants of KCNJ10 are responsible for drug resistance. Guo Y, et al. [15] proposed that the rs12402969 genotype of the KCNJ10 gene is responsible for AED resistance . However, Zhu H, et al. [16] assessed relationship of nine loci of the KCNJ10 gene (rs12122979, rs1186685, rs6690889, rs2486253, rs1186675, rs12402969, rs12729701, rs1890532 and rs3795339) with ASMs resistance and could not find any relationship between these genotypes and drug resistance . It seems that investigations should focus on the detection of responsible genotypes for ASMs resistance epilepsies.
Different responsible genes in drug- resistant epilepsy detected. These include sodium voltage-gated channels SCN1A, SCN2A, SCN3A, SCN8A and SCN1B); potassium voltage-gated channels (KCNA1, KCNA2, KCNB1, KCND7, KCNH5, KCNJ10, KCNQ2 and KCNT1); calcium voltage-gated channel subunit α1 H; mitochondrial transporter family members (SLC2A1, SLC6A1, SLC6A4, SLC6A11, SLC9A6, SLC25A22 and SLC35A2); aTP binding-cassette transporters (ABCB1, ABCC2, ABCC5 and ABCG2); drug-metabolizing enzymes (CYP2C1, CYP2C9, CYP2C19, CYP2D6, CYPP3A4 and CYP3A5); cdKl5; and GaBa receptors (GABRA1 and GABBR1). We also found SCN1A, SCN2A, and SCN8A as sodium voltage- gated channels, KCNJ10 as potassium voltage-gated channels, and SLC6A1 and SLC25A22, as mitochondrial transporter family. However, these mutations not confined to only the above- mentioned classification.
Moreover, the vast number of detected genes with different underlying mechanism for drug- resistant epilepsy makes the fact of “percision medicine” difficult. we are still far from precision medicine. In our investigation, only 38.46% of the patients had complete responses to ASMs changes. However, still 13.46% had no response and 19.23% had a weak response.
Our study limited by the retrospective assessment, and therefore, we could not assign many related factors to precision medicine. However, the assessment of 52 confirmed drug- resistant epilepsy cases is a fact that barely happened in other similar studies. We also proposed various mutated genes that can be helpful for the researcher and clinicians in this field.
Author’s Contribution
Farajirad Elnaz conceived and supervised the data; Mirzadeh Hs prepared the first draft of the paper; Tohidi H edited the final proof, Ashrafzadeh F, Akhondian J, Beiraghi- Toosi M, Imannezhad Sh, and Hashemi N referred patients and searched for relevant articles, Ghayoor Karimiani E assessed genetic studies. All authors discussed the results and contributed to the final manuscript.
Conflict of Interest
Authors declare no conflict of interest.
Aknowlegment
We would like to acknowledge all the parents’ of the children who helped the conduction of this study.
References
-
Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Hauser WA, et al. (2010) Definition of drug resistant epilepsy: Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 51(6): 1069-1077.
-
Xue-Ping W, Hai-Jiao W, Li-Na Z, Xu D, Ling L (2019) Risk factors for drug-resistant epilepsy: A systematic review and meta-analysis. Medicine 98(30): e16402.
-
Strzelczyk A, Griebel C, Lux W, Rosenow F, Reese JP (2017) The Burden of Severely Drug-Refractory Epilepsy: A Comparative Longitudinal Evaluation of Mortality, Morbidity, Resource Use, and Cost Using German Health Insurance Data. Frontiers in Neurology 22: 8: 712.
-
Kumar G (2021) Evaluation and management of drug resistant epilepsy in children. Current Problems in Pediatric and Adolescent Health Care 51(7): 101035.
-
Mangunatmadja I, Indra RM, Widodo DP, Rafli A (2021) Risk Factors for Drug Resistance in Epileptic Children with Age of Onset above Five Years: A Case-Control Study. Behavioural Neurology 2021: 9092824.
-
Cárdenas‑Rodríguez N, Carmona‑Aparicio L, Pérez‑Lozano DL, Ortega‑Cuellar D, Gómez‑Manzo S, et al. (2020) Genetic variations associated with pharmacoresistant epilepsy. Molecular Medicine Reports 21(4): 1685-1701.
-
Carvill GL, Engel KL, Ramamurthy A, Cochran JN, Roovers J, et al. (2018) Aberrant inclusion of a poison exon causes dravet syndrome and related SCN1A-associated genetic epilepsies. The American Journal of Human Genetics 103(6): 1022-1029.
-
Wong JC, Shapiro L, Thelin JT, Heaton EC, Zaman RU, et al. (2021) Nanoparticle encapsulated oxytocin increases resistance to induced seizures and restores social behavior in Scn1a-derived epilepsy. Neurobiology of disease 147: 105147.
-
Myers KA, Shevell MI, Sébire G (2019) Sudden unexpected death in GEFS+ families with sodium channel pathogenic variants. Epilepsy Research 150: 66-69.
-
Nazish HR, Ali N, Ullah S (2018) The possible effect of SCN1A and SCN2A genetic variants on carbamazepine response among Khyber Pakhtunkhwa epileptic patients, Pakistan. Ther Clin Risk Manag 14: 2305-2313.
-
Usluer S, Salar S, Arslan M, Yiş U, Kara B, et al. (2016) SCN1A gene sequencing in 46 Turkish epilepsy patients disclosed 12 novel mutations. Seizure 39: 34-43.
-
Angelopoulou C, Veletza S, Heliopoulos I, Vadikolias K, Tripsianis G, et al. (2017) Association of SCN1A gene polymorphism with antiepileptic drug responsiveness in the population of Thrace, Greece. Archives of Medical Science 13(1): 138-147.
-
Reid ES, Williams H, Anderson G, Benatti M, Chong K, et al. (2017) Mutations in SLC25A22: hyperprolinaemia, vacuolated fibroblasts and presentation with developmental delay J Inherit Metab Dis 40(3): 385-394.
-
Shbarou R (2016) Current treatment options for early- onset pediatric epileptic encephalopathies. Current treatment options in neurology 18(10): 44.
-
Guo Y, Yan KP, Qu Q, Qu J, Chen ZG, et al. (2015) Common variants of KCNJ10 are associated with susceptibility and anti-epileptic drug resistance in Chinese genetic generalized epilepsies. PLoS One. 10(4): e0124896.
-
Zhu H, Zhang M, Fu Y, Long H, Xiao W, et al. (2020) Effects of AQP4 and KCNJ10 gene polymorphisms on drug resistance and seizure susceptibility in Chinese Han patients with focal epilepsy. Neuropsychiatric Disease and Treatment 16: 119-129.
- Management of Chronic Insertional Achilles Tendinopathy Using Flexor Hallucis Longus Tendon Transfer in Patients Over 50 Years of Age: A Four-Case Series Following the CARE Guidelines
- Application of Induced Pluripotent Stem Cells in Bone Tissue Engineering: Current Status and Prospects
- Surgical Management of Upper Thoracic Esophageal Squamous Cell Carcinoma with Concomitant Hypersplenism: Integration of Chai's Supra-Thoracic Apex Technique with Laparoscopic Splenectomy - A Technical Innovation Case Study with Systematic Review
- Evaluation of Masticatory Functional Efficiency of Stomatognathic System in Patients Undergoing Open Reduction Internal Fixation for Treatment of Pan-Facial Trauma: A Prospective Study
- Hepatic Abscess Secondary to Appendiceal Phlegmon an Unusual Complication of Appendiceal Phlegmon
- Report of Lumboperitoneal (LP) Shunt Procedure in Over Decades Experiences, Systematic Narrative Review