Clinical Case Reports 1971-1974
In this paper five case reports were presented and include congenital heart disease in de Lange Syndrome, management of growth retardation in steroid dependent asthma, systemic venous anomalies and partial heterotaxia with normal heart, Ebstein’s malformation of the tricuspid valve with atresia, and superior vena caval obstruction in total anomalous pulmonary venous connection. For each case report, clinical, chest x-ray, electrocardiogram and other pertinent findings were presented. This was followed by discussion of etiology, diagnosis, and treatment options, as appropriate.
Introduction
During the academic clinical practice for over five decades, the author had the unique opportunity to observe and document many interesting clinical case scenarios. The purpose of this review is to revisit these interesting cases. Because of the voluminous amount of this material, the material may be divided into several parts. In the first paper of this series five cases seen/documented in 1970 were presented [1]. In this paper case seen 1971-1974 will be reviewed. Each of these case reports, while rare and important clinical observations, do demonstrate a clinical point that is useful to the pediatricians, pediatric cardiologists and/or other physicians.
Congenital Heart Disease in the De Lange Syndrome
Introduction: The prevalence of congenital heart disease (CHD) in de Lange syndrome has not been systematically studied as of early 1970s. The objective of our report [2] was to present a patient with classic manifestations of de Lange syndrome with pulmonary stenosis and to examine the literature to determine the incidence and types of CHD in this syndrome.
feet were also obvious.
Physical examination at two week of age revealed right ventricular heave, normal first heart sound, split second heart sound with diminished pulmonary component, a grade 3/6 ejection systolic murmur heard best at the upper left sternal border with radiation into the back bilaterally, no diastolic murmurs, and no signs of congestive heart failure (CHF).
Chest x-ray revealed 13 ribs on both sides, 2 mid- thoracic hemi-vertebrae, and an odd segmental pattern of the sternum, mild cardiac enlargement, and normal pulmonary vascular markings. Upper extremity radiographs revealed short humerus and a single forearm bone, probably radius, on both sides. The electrocardiogram (ECG) indicated right axis deviation and right atrial (RA) and right ventricular (RV) hypertrophy. Normal male karyotype was found on chromosomal analysis. Cardiac catheterization and cineangiography performed at 3 weeks of age confirmed the diagnosis of severe valvar pulmonary stenosis with intact ventricular septum, a small patent ductus arteriosus (PDA) and a patent foramen ovale (PFO). The baby was transferred to another hospital for palliative care.
Literature Review: Detailed review of the literature identified 250 cases of de Lange syndrome from 117 papers [1]. These papers were examined for the presence or absence of CHD and if present, the type of CHD. Three groups were identified. Group I was made up of patients with a definitive diagnosis of CHD - 37 of 250 (15%). Group II consisted of
30 (12%) cases in whom physical findings, usually a cardiac murmur, suggestive of CHD were present, but no additional finding to confirm CHD were presented by the respective authors. Group III patients had no CHD. The type of heart defect identified is listed in Table 1; ventricular septal defect (VSD) appears to be the most common defect.
| Nature of heart defect | Number |
|---|---|
| Ventricular septal defect | 10 |
| Patent ductus arteriosus | 7 |
| Pulmonary stenosis* | 6 |
| Patent foramen ovale | 5 |
| Anomalous systemic venous drainage | 3 |
| Atrial septal defect | 3 |
| Tetralogy of Fallot | 2 |
| Miscellaneous** | 18 |
| Total | 54# |
*Includes one case with infundibular pulmonary stenosis and one with a bicuspid pulmonary valve. One case each of truncus arteriosus, aortic stenosis, hypoplasia of one of the cusps of aortic valve, coarctation of aorta, right aortic arch, separate origin of the right subclavian and carotid arteries from aortic arch, hypoplastic left heart syndrome, and high origin of right coronary artery. Two cases each of abnormal insertion of chordae tendineae and endocardial fibroelastosis in association with other defects. In six cases the defect was not clearly stated or not stated. #The discrepancy between totals is due to the occurrence of more than one defect in some patients. Table 1**: Relative incidence of various known types of congenital heart defects in de Lange syndrome (37 cases).
Discussion: The baby presented had typical features of de Lange syndrome. Cardiac findings were those of valvar pulmonary stenosis, confirmed by cardiac catheterization and cine-angiography. Extensive review of previously published reports indicated at least 15% prevalence of CHD, significantly higher than 0.8% incidence in normal population. The incidence of CHD is lower than that seen with Down syndrome, and we concluded that CHD is not an integral part of de Lange syndrome [1]. However, it should be noted that many children (at least 12%) had not been adequately studied to determine the presence and type of CHD. Consequently, the true incidence of CHD may be higher. The most common defect in de Lange syndrome in this survey was VSD, like that seen normal population.
Because of invariably severe mental retardation associated with this syndrome, no open heart surgery to treat pulmonary stenosis was undertaken, in keeping with thinking in 1970s. However, more recent de Lange syndrome cases seen and cared for by the author did undergo balloon pulmonary valvuloplasty to treat valvar pulmonary stenosis.
Growth Retardation in Steroid Dependent Asthma-Management with Corticotrophin (ACTH)
Introduction: Several complications have been reported with long-term therapy using steroids in children. More common among these complications are adrenal atrophy with failure to react in stressful situations, suppression of obvious signs of infection, and growth failure [3]. The purpose of our paper was to document growth retardation in a patient who was on long-term prednisone treatment who was switched to corticotrophin (ACTH) in an attempt restore his growth [3].
Case Report: A 12-1/2 year-old young man was seen for evaluation and management of his asthma. He had a perennial asthma from early infancy. He was skin tested and desensitized for house-dust, molds pollens and bacteria multiple times with only transient improvement with each of these regimens. In addition to conventional bronchodilator therapy available at that time, he required oral prednisone, 5 to 10 mg daily for control of his asthma. When evaluated by us at the age of 12-1/2 years, his heart and respiratory rates were normal, the antero-posterior diameter of the chest was markedly increased, and auscultation revealed moderate expiratory wheezes and many scattered rhonchi bilaterally.
Chest roentgenogram revealed marked hyperinflation with flattened diaphragms. Pulmonary function studies were consistent with striking obstructive airway disease which reversed partially following bronchodilators. He was managed with a program that included environmental control, oral and aerosol bronchodilators, and maintenance doses of 5 to 10 mg of prednisone per day. His symptoms were under good control, with an infrequent need for subcutaneous sympathomimetic medications. Efforts to modify prednisone therapy to an alternate day regimen failed. The most important problem was the absence of growth (Figure 2) during the two and a half years of treatment he had grown only 3.0 cm, an obviously decreased growth velocity (Figure 2A) and remarkably reduced standing height (Figure 2B).
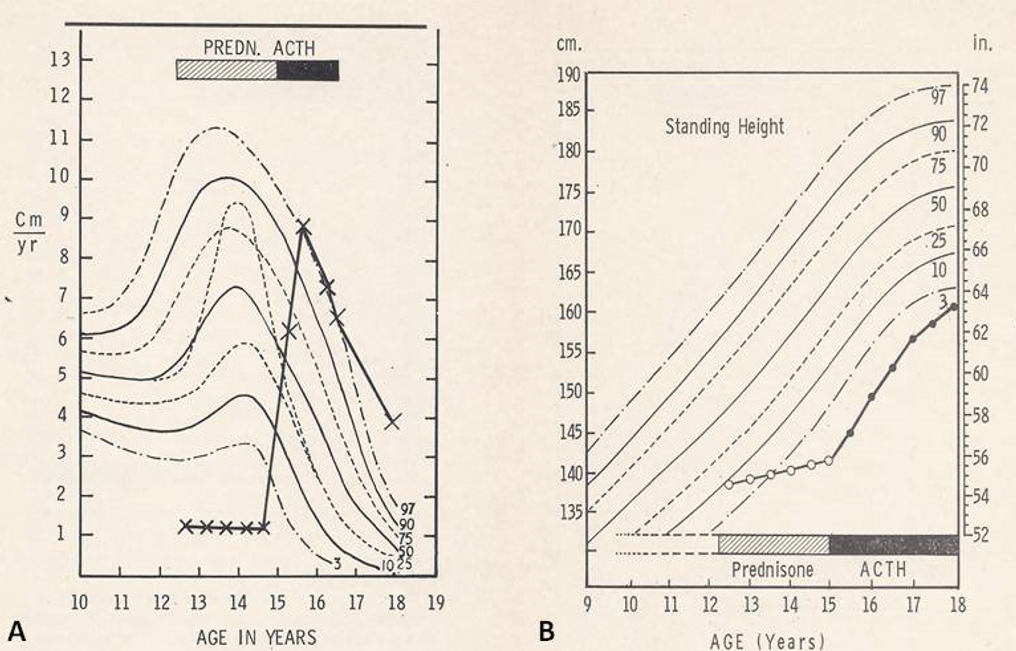
Figure 2A: Patient’s height velocity was plotted on height velocity curve from Tanner, et al. [4]. Normal percentiles (3rd through 97th) of height velocity are marked. The patient’s height velocity was calculated as cm/yr. per every six month period of observation. Prednisone (hashed bar) and ACTH (solid bar) periods are shown. The height velocity was below 3rd percentile during the prednisone treated period while the height velocity varied from 50th to above 97th percentile during ACTH treatment, a remarkable improvement. B. This is similar to A, but patient’s standing height was plotted on curve from Tanner, et al. [4]. Normal standing height percentiles (3rd through 97th) for age are drawn. Again, prednisone and ACTH period are indicated. Growth failure while on prednisone therapy and improvement with ACTH was clearly noted. Reproduced from Rao PS, Lipow HW. Clin Pediat 1972; 11:93-97.
To address this growth problem, his treatment was switched to daily subcutaneous injections of 40 units of ACTH at 15 years of age. During the following year and a half, he grew 11.5 cm. His height velocity has improved and normalized (Figure 2A) and his standing height increased (Figure 2B). The bone age was 11.5 years prior to starting ACTH treatment and advanced to 14 years after the one and a half years of ACTH therapy. Prior to ACTH he had no secondary sexual characteristics, but growth of pubic hair started after year and a half treatment with ACTH. During the period of ACTH therapy, the control of the asthma was like the time of prednisone treatment regarding the need for bronchodilators, hospitalizations, and number of days absent from the school. Pulmonary function tests after a year and a half of ACTH therapy showed mild obstruction with hyperinflation. After one and one-half years, his ACTH administration was reduced to 40 units on alternate days. By the end of the period of observation when he was 18 years old, his linear growth was satisfactory (Figure 2) with a height of 63-1/4 inches placing him out of the significantly dwarfed range.
Discussion: Inhibition of growth with long-term treatment with steroids is well recognized [3] and our patient is a good example of such a phenomenon. Growth retardation associated with steroid treatment of asthma, nephrotic syndrome and rheumatoid arthritis appears to improve by substitution of prednisone with ACTH or a combination of ACTH and steroids [4], as demonstrated in our patient. The mechanism for growth retardation with steroids is not clearly understood but is likely to be suppression of endogenous ACTH by prednisone. When ACTH is administered, it may stimulate adrenals to produce growth-promoting factors. Several practical tips regarding the usage of steroids in children were reviewed for the interested reader [3]. In summary, when long-term steroid treatment is necessary in children during the growth period, their height should be closely monitored. If growth retardation is documented, switching to ACTH should be entertained.
Systemic Venous Anomalies and Partial Heterotaxia with Normal Heart
Introduction: Partial heterotaxia and systemic venous anomalies are usually associated with complex CHDs. The author presented a 5-year-old child with incomplete visceral inversion (partial heterotaxia) (Figure 3) and systemic venous anomalies (Figures 4 & 5) without any associated CHD [5].
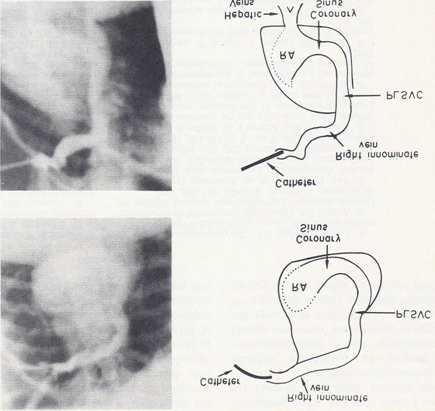
Figure 4: Selected frames from right innominate vein cine-angiograms in postero-anterior (top) and left anterior oblique (bottom) projections, demonstrating the connection of the right innominate vein to a persistent left superior vena cava (PLSVC) which empties into the coronary sinus and right atrium (RA). The hepatic veins seem to enter the RA directly. Also note the absence of the right superior vena cava. The line drawings on the right are provided to improve the clarity. Reproduced from Rao PS, Molthan ME, Am J Dis Child 1973; 125:749-52.
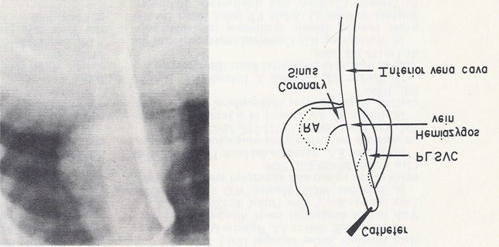
Figure 5: Selected frame from inferior vena caval cine-angiogram in postero-anterior projection, demonstrating the connection of the inferior vena cava to the hemiazygos vein which empties into to a persistent left superior vena cava (PLSVC), which in turn empties into the coronary sinus and right atrium (RA). The line drawing on the right are provided to improve the clarity. Reproduced from Rao PS, Molthan ME, Am J Dis Child 1973; 125:749-52.
Case Report: This 5-year-old girl had ejection systolic and mid-diastolic murmurs on physical examination, a P vector (axis) of -60° on ECG and mesocardia on a chest x-ray (Figure 6) with the appendix on the left side (Figure 3), prompting cardiac catheterization. No Howell-Jolly bodies were found upon blood smear. The liver/spleen and stomach bubble and tracheo-bronchial tree pattern appeared to be normal, consistent with visceral situs solitus.
Cardiac catheterization data revealed normal pressures in all heart chambers, without any evidence of shunts. However, the catheter course was abnormal. This was related to a persistent left superior vena cava entering the right atrium via an enlarged coronary sinus (Figure 4), an absent right superior vena cava (Figure 4) and an infra-hepatic interruption of the inferior vena cava with hemiazygos continuation into the persistent left superior vena cava (Figure 5). The hepatic veins seemed to enter the right atrium directly (Figure 4). No other intra-cardiac abnormalities were noted.
Discussion: The emptying of most of the systemic venous return into the right atrium may have caused the diastolic murmur and the systolic murmur and was thought to be a functional murmur. In this case with partial heterotaxia and systemic venous anomalies, the heart was otherwise normal, which is unusual and prompted us to document the case because of prior reported association of complex heart disease with systemic venous anomalies and partial inversion of the abdominal viscera, as quoted in our paper [6]. The normal position of the atria may have prevented the development of a complex CHD, despite the systemic venous anomalies [6]. The possible association of the described partial heterotaxia and systemic venous anomalies with asplenia/polysplenia syndromes was discussed [6]. Other clinical applications of these findings were explored, including the association of the superior vector of the P wave with vena caval anomalies, the difficulties in advancing the catheters into the cardiac chambers in the presence of these vena caval anomalies, and the surgical implications [6].
Ebstein’s Malformation of the Tricuspid Valve with Atresia
Introduction: We have observed a rare case of Ebstein’s anomaly of the tricuspid valve with atresia of the tricuspid orifice; this child died following an attempted palliative surgical procedure at the age of two years [7]. The clinical course, ECG findings, catheterization and angiographic data, and pathological features of this case were presented. The differentiation of this anomaly from classic cases of tricuspid atresia was also discussed [7].
Case Report: An 8 1/2 lb. weighing male infant was born following a full-term normal pregnancy and delivery. A cardiac murmur was detected on day one of life. He presented with signs of CHF at the age of four weeks. Physical examination showed tachycardia, tachypnea, and mid cyanosis. The blood pressure and pulses were normal. A thrill was felt at the lower left lower sternal margin. On auscultation, a normal first heart sound and a single second heart sound were heard. There were two murmurs: a grade 4/6 holosystolic murmur at the lower left sternal border and a grade 2/6 mid- diastolic flow murmur at the apex. The liver was felt four cm below the right costal margin and the spleen, one cm below the left costal margin.
ECG and vectorcardiogram demonstrated a left axis deviation with a mean frontal plane vector of -40°, right atrial enlargement and left ventricular hypertrophy. Chest x-ray displayed moderate cardiomegaly with right atrial enlargement and increased pulmonary blood flow.
Hemoglobin was 11.5 g/100ml. Cardiac catheterization at the age of nine weeks revealed right to left shunt at atrial level with arterial desaturation into low 80s and mildly elevated right atrial pressures. Left ventricular cine-angiogram (Figure 7A) demonstrated a moderate-sized ventricular septal defect. The right atrial angiogram in the postero- anterior view showed opacification of the left atrium and left ventricle before opacification of the right ventricle, a feature like that seen in classic case of tricuspid atresia. Detailed examination of right atrial angiogram (Figure 7B) revealed that the right atrial border is rounded and extends more to the left than seen in other cases of tricuspid atresia. A close- up view is shown in Figure 8. The baby improved following CHF therapy with digoxin and diuretics.
![Figure 7: Selected left ventricular (LV) cine-angiographic frame in left anterior oblique view demonstrating a ventricular septal defect (VSD). The right ventricle (RV) is opacified via the VSD and looks close to normal in size. b. Selected right atrial (RA) cine-angiographic frame in postero-anterior view prior to opacification of the left atrium (LA). The contrast material delineates the atretic tricuspid valve and is shown by small unlabeled arrows. This is rounded and extends more to the left than in other tricuspid atresia cases. c and d. Right atrial angiographic frames from two patients with classic tricuspid atresia are shown for comparison with figure b. These frames are frozen prior to full opacification of the LA. PA, pulmonary artery; RAA, right atrial appendage; SVC, superior vena cava. Reproduced from Rao PS, et al. [7].](/fulltextimages/11428/fig_7.png)
Figure 7: Selected left ventricular (LV) cine-angiographic frame in left anterior oblique view demonstrating a ventricular septal defect (VSD). The right ventricle (RV) is opacified via the VSD and looks close to normal in size. b. Selected right atrial (RA) cine-angiographic frame in postero-anterior view prior to opacification of the left atrium (LA). The contrast material delineates the atretic tricuspid valve and is shown by small unlabeled arrows. This is rounded and extends more to the left than in other tricuspid atresia cases. c and d. Right atrial angiographic frames from two patients with classic tricuspid atresia are shown for comparison with figure b. These frames are frozen prior to full opacification of the LA. PA, pulmonary artery; RAA, right atrial appendage; SVC, superior vena cava. Reproduced from Rao PS, et al. [7].
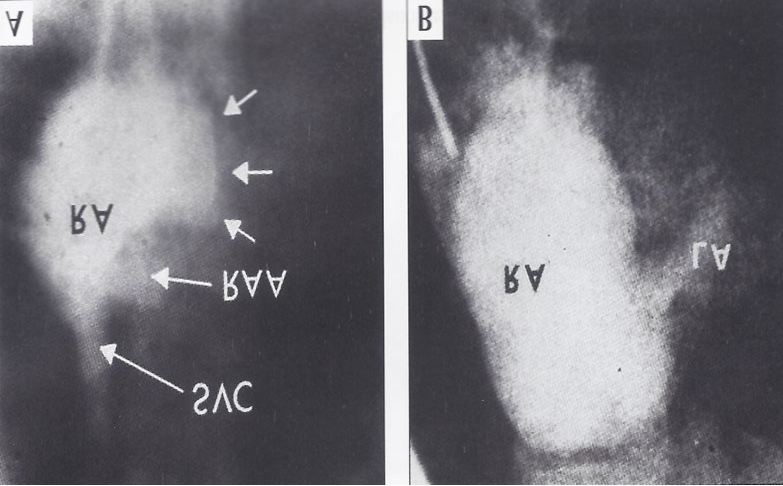
Figure 8A,8B & Figure 7B,7C are put together to illustrate right atrial angiographic differences more clearly between Ebstein’s and classic types of tricuspid atresia. Abbreviations are same as those used in Figure 7. Reproduced from Rao PS, et al. [7].
The child was re-hospitalized at 2 years of age because of edema, hepatomegaly, and easy fatigability. Findings on physical examination were like those of the previous visit except for hepatomegaly, 6 cm below the right costal margin and increased pulsatility. Hemoglobin was elevated at 19.6 g/100 ml. ECG, vectorcardiogram and chest x-ray were also like previous visit. Cardiac catheterization was repeated which showed a mean pressure gradient of 6.5 mmHg across the atrial septum and reduced left ventricular oxygen saturation. Otherwise, these findings were like those of prior catheterization. Findings on angiography were also like those of the initial catheterization and are consistent tricuspid atresia diagnosis.
To relieve the interatrial obstruction, atrial septectomy was performed under inflow occlusion. Following the procedure, the patient sustained a cardiac arrest and resuscitation efforts were unsuccessful. Postmortem examination revealed an enlarged heart with normally related great vessels. Enlarged RA and wide-open surgically made atrial septal defect were seen. Tricuspid valve leaflets were plastered onto the right ventricular wall akin to Ebstein’s anomaly (Figures 9A,9B) and their edges were fused with no tricuspid valve opening, like tricuspid atresia. No clear-cut commissures or raphes were seen. Mild enlargement of the left atrium and left ventricle was observed, but the aorta was normal. A small, slit-like VSD was present. The pulmonary valve and the aortic valve were near normal in size. The RV outflow tract was mildly hypoplastic. The RV inflow tract was also slightly small. However, if the atrialized portion of the RV is included, the RV size appeared sufficient to support normal pulmonary circulation. The main pulmonary trunk and the branches appeared normal. There was no patency of the ductus arteriosus.
![Figure 9: The interior of the right atrium and the atrialized right ventricle (ARV) are shown. B. This is close-up of A. The interrupted line demarcates the true tricuspid valve annulus. The tricuspid valve (TV) leaflets are displaced downward (Ebstein’s malformation) and fused, incorporating a part of the right ventricle into the right atrium (RA). FO, fossa ovalis. Reproduced from Rao PS, et al. [7].](/fulltextimages/11428/fig_9.png)
Figure 9: The interior of the right atrium and the atrialized right ventricle (ARV) are shown. B. This is close-up of A. The interrupted line demarcates the true tricuspid valve annulus. The tricuspid valve (TV) leaflets are displaced downward (Ebstein’s malformation) and fused, incorporating a part of the right ventricle into the right atrium (RA). FO, fossa ovalis. Reproduced from Rao PS, et al. [7].
Discussion: The clinical, ECG (abnormal superior vector [left axis deviation]) and hemodynamic data were indistinguishable from tricuspid atresia. The catheter course during cardiac catheterization was also like that seen in classic tricuspid atresia. A left ventricular cine-angiogram revealed opacification of the RV via a VSD (Figure 7A), again, similarly to tricuspid atresia. RA angiography revealed a sequential opacification of the left atrium and left ventricle, which was also similar to that seen in classic tricuspid atresia. However, the contrast material outlining the atretic tricuspid valve was rounded and extended more to the right (Figures 7B & 8A) than is seen in classic tricuspid atresia cases, and was distinctly different from that visualized in classic tricuspid atresia (Figures 7C,7D). A focused view of the figures 7b and c is shown in Figure 8 to improve the clarity. The pathological features are distinctive (Figure 9) in that all three leaflets of the tricuspid valve were displaced downward and plastered onto the right ventricular wall, and a significant proportion of the right ventricle was atrialized.
The rarity of this disease entity was pointed out; it forms 2.5% to 8.0% of tricuspid atresia cases, and 3.6% of Ebstein’s malformation of the tricuspid valve cases. It was also stated that this case represents the fourth case recorded up to that time; the first three cases were those described by Van Praagh and his associates, though in an abstract form [8]. We have briefly discussed embryology and nomenclature and concluded that this lesion belongs in the Ebstein’s malformation of the tricuspid valve group, rather than in the tricuspid atresia group, but conceded that this condition may be categorized with the tricuspid atresia group, especially when physiologic and diagnostic considerations are involved. Discussion of a surgical approach followed: we suggested excision of the tricuspid valve, replacing it with a prosthetic valve and excluding the atrialized portion of the right ventricle with or without leaving open an interatrial communication [7]. The surgical approach is distinctly different from that used for classic tricuspid atresia (palliative systemic to pulmonary shunt, preparatory to future Fontan).
Superior Vena Caval Obstruction in Total Anomalous Pulmonary Venous Connection
Introduction: Total anomalous pulmonary venous connection (TAPVC) is generally classified into supra- diaphragmatic and infra-diaphragmatic types. Infra- diaphragmatic type is typically associated with obstruction to pulmonary venous return while the supra-diaphragmatic type is rarely associated with pulmonary venous obstruction. The objective of this report was to present an atypical type of obstruction to pulmonary venous return in a case of supra-diaphragmatic TAPVC and to review the literature to determine types of obstruction seen in the supra- diaphragmatic TAPVC [9].
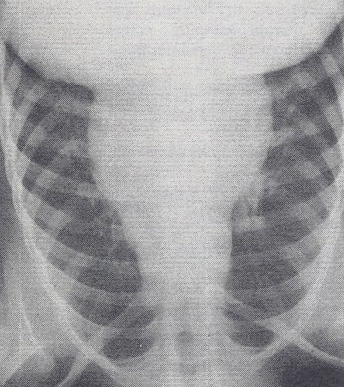
Case Report: A male infant, a product of a full-term normal pregnancy and delivery, presented with signs of CHF at the age of 2 months. Examination revealed tachypnea, mild to moderate cyanosis, a heart rate of 180/minute, normal blood pressure, and an enlarged liver, palpable 5 cm below the right costal margin. The precordium was hyper-dynamic with a prominent RV impulse. The second heart sound was narrowly split. A grade 2/6 ejection systolic murmur at the left upper sternal border, and a grade 2/6 mid-diastolic flow rumble at the left lower sternal border were heard. Chest roentgenogram showed moderate cardiomegaly with increased pulmonary vascular markings (Figure 10). The ECG showed RA and RV hypertrophy (Figure 11).
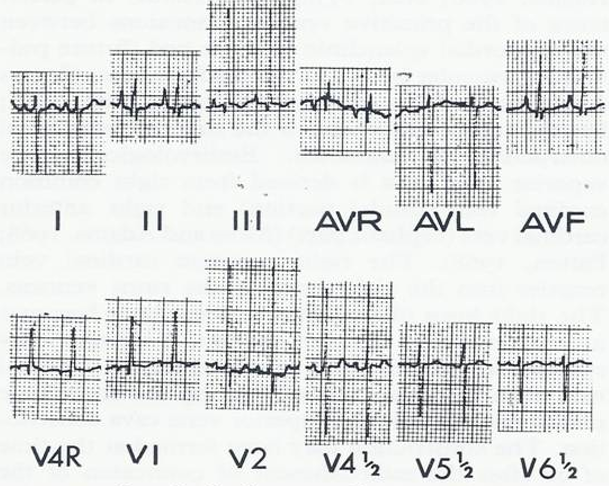
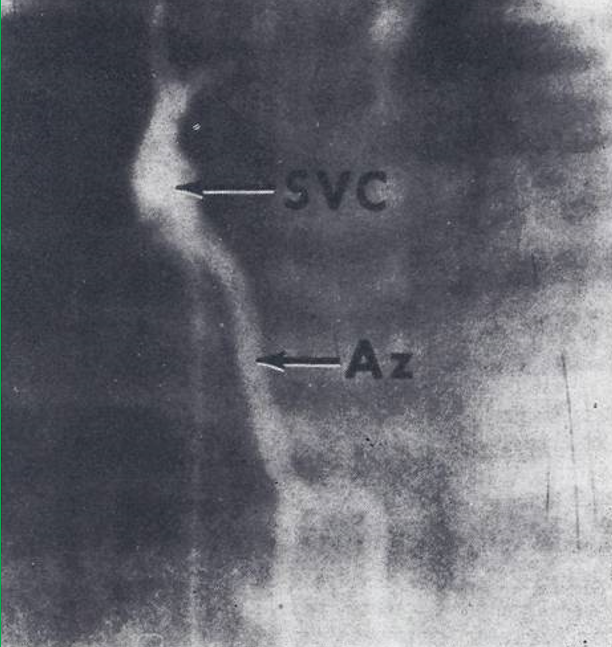
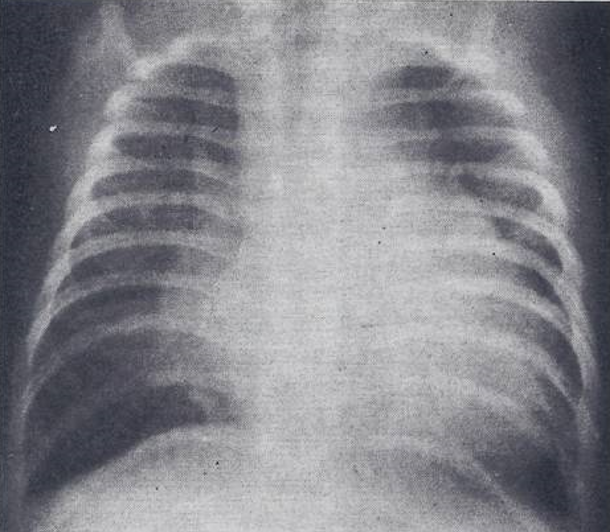
Figure 12: Selected frame from superior vena caval (SVC) cine-angiogram in posteroanterior view showing prompt opacification of the azygos vein (Az). Note absence of opacification of the right atrium. In the cine-angiographic frames that followed the inferior vena cava was filled with contrast subsequent to Az opacification and then the right atrium. Reproduced from Rao PS, Silbert DR. Brit Heart J 1974; 36:228-32.
Following treatment of CHF, cardiac catheterization and selective cine-angiography were carried out. The RV and pulmonary artery (PA) pressures were at systemic level.
There was a 12 mmHg mean pressure difference between the right atrium and the superior vena cava (SVC). Levophase of the main pulmonary artery cine-angiogram was faint without opacification of a common pulmonary vein. But the SVC was opacified faintly with subsequent RA opacification. SVC cine-angiogram (Figure 12) showed retrograde filling of the azygos vein with subsequent opacification of inferior vena cava and RA. Only a tiny amount of contrast passed from the SVC into the right atrium.
Attempts to catheterize the common pulmonary vein from the SVC failed, and therefore, additional pressure gradient from the common pulmonary vein to the SVC could not be excluded. Balloon atrial septostomy was performed which abolished the interatrial pressure gradient (2.0 mmHg vs. 0.5 mmHg). Treatment of CHF was continued. Since there was persistent CHF and failure to gain weight, surgical relief of the SVC obstruction was undertaken at the age of 5 months. At surgery, the SVC appears to be inserted into the right atrium in a more posterior position than is normally seen. A linear vertical incision was made at the RA and SVC junction which was closed in a transverse fashion. The pressures in the SVC and right atrium were equalized after the procedure. The infant seemed to improve, but the next morning the baby suddenly deteriorated and died.
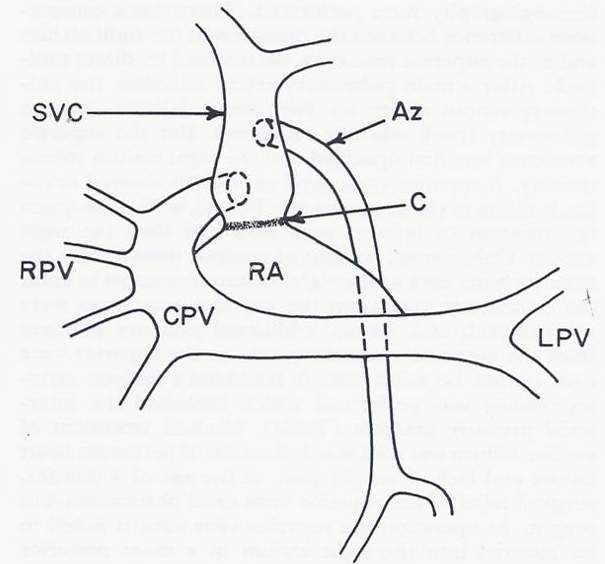
Figure 13: Diagrammatic sketch of the pulmonary venous return found on postmortem examination. Two pulmonary veins drain the left lung fuse and form a common pulmonary vein (CPV) which traverses to the right and then ascends to empty into the postero-lateral aspect of the superior vena cava (SVC). Four right pulmonary veins (RPVs) join the lateral convexity of the CPV. The position of the azygos vein (Az) opening into the SVC and the constricted site of the SVC (arrow) are also shown. C, constricted area of the SVC; LPV, left pulmonary veins; RA, right atrium. Reproduced from Rao PS, Silbert DR. Brit Heart J 1974; 36:228-32.
Autopsy findings revealed RV and PA enlargement. The left atrium and the left ventricle were of normal size. The pulmonary venous drainage is diagrammatically portrayed in Figure 13. Two pulmonary veins drain the left lung which joined each other behind the heart. This common pulmonary vein coursed across the midline and ascended superiorly to join the postero-lateral aspect of the SVC. Four pulmonary veins from the right lung joined the lateral convexity of the common pulmonary vein (Figure 13). The common pulmonary vein emptied into the SVC; its orifice was located slightly inferior to the opening of the azygos vein. The SVC was of adequate size, but there was a ring-like fibrous constriction at the junction of the SVC with the RA which was partially enlarged during surgery. On histological examination grade II changes (Heath-Edwards) of the pulmonary vasculature were seen.
Literature Review
A detailed review of the literature was performed to discern the types of obstruction seen in supra-diaphragmatic TAPVC [9]. This review indicated that pulmonary venous obstruction occurs in all types of supra-diaphragmatic TAPVC and each of these was referenced in our publication [9]. In the most common type of TAPVC, connection to the left innominate vein, the obstruction of the vertical vein may be intrinsic or extrinsic. The intrinsic obstruction can result from narrowing of the anomalous vertical vein at its junction with the common pulmonary vein or at its junction with the left innominate vein or it may be due to narrowing of the left innominate vein. The extrinsic obstruction is secondary to compression of the vertical vein as it passes between left pulmonary artery and left bronchus. In cases with connection of common pulmonary vein with the azygos vein, the connecting vessel was small causing obstruction in one case and in another case the nature of the obstruction was not described [9]. In cases with connection to the RA, atresia or stenosis of the vessel connecting the common pulmonary vein to the RA can occur. In subjects with connection to the coronary sinus, stenosis at the junction of the pulmonary venous confluence with the coronary sinus and stenosis of the individual pulmonary veins as they enter the coronary sinus has been reported. In patients with connection to the azygos vein, small or narrow connecting vessel was present causing obstruction. Obstruction may occur at the level of the atrial septum which could be treated with balloon atrial septostomy. Rarely, the common pulmonary vein may not have any connection with the systemic veins; such cases are not truly TAPVCs [9].
In summary, our review indicated that pulmonary venous obstruction occurs more frequently in supra- diaphragmatic types of TAPVC than was thought prior to the review. The obstruction to pulmonary venous return in cases with TAPVC may be classified into
- intrinsic stenosis or atresia of the vessel connecting the common pulmonary vein with systemic venous system,
- extrinsic compression of the connecting vessel and
- obstruction at the atrial septal level [9].
Discussion: Our case is a rare instance of obstruction in supra-diaphragmatic TAPVC with physiologically and anatomically confirmed obstruction at the junction of the SVC with the RA. At the time of that report, only few other similar cases were reported in the literature, one had stenotic ring in the SVC like our case and other cases had stenosis of the common pulmonary vein at its entrance into the SVC [9]. The other types obstructed supra-diaphragmatic TAPVC described as of the time our report was reviewed in the preceding literature review section. The embryology of TAPVC with particular attention to the development of circular constriction of the SVC was reviewed [9] for the interested reader. In summary, we presented an unusual case of supra-diaphragmatic TAPVC in which physiologically and anatomically proven stenosis in the SVC was documented. Extensive review of the literature indicated that pulmonary venous obstruction occurs in supra-diaphragmatic TAPVC more often than previously recognized [9].
Summary and Conclusion
In this report, multiple case scenarios were presented. In the first report, CHDs seen in patients with de Lange syndrome were reviewed. Because of severe mental retardation seen in this syndrome, open-heart surgery is not generally recommended; however, transcatheter intervention may be performed for suitable lesions such as pulmonary stenosis. In children with severe asthma, steroids (prednisolone) may become necessary to control symptoms, but such treatment invariably produces growth retardation. In such cases, switching treatment using ACTH may avert growth problems. Partial heterotaxia and systemic venous anomalies are frequently seen in association with complex CHDs. Detailed clinical, radiological, and angiographic features of a 5-year-old child with partial heterotaxia and systemic venous anomalies who had no associated CHD was presented. This unusual association of systemic venous anomalies and partial inversion of the abdominal viscera with otherwise normal heart was pointed out. In the fourth case, angiographic and pathologic features of a rare combination of Ebstein’s anomaly of the tricuspid valve with atresia was presented. We have suggested that tricuspid valve excision along with replacing it with a prosthetic valve as a surgical approach instead of Fontan for these types of cases. In the final case, the association of pulmonary venous obstruction in a patient with supra-diaphragmatic TAPVC was illustrated. It is generally known that pulmonary venous obstruction is classically present in the infra-diaphragmatic types of TAPVC while it is absent in supra-diaphragmatic types. The presented case is an exception to this rule; review of the literature revealed that obstruction to pulmonary venous return in the supra-diaphragmatic types is more often present than is recognized. The cases presented illustrate different clinical scenarios that may be useful to the clinician.
References
-
Rao PS (2023) Clinical Case Reports-1970. Jour of Clin Cas Rep 5(5).
-
Rao PS, Sissman NJ (1971) Congenital heart disease in the de Lange syndrome. J Pediatr 79(4): 674-677.
-
Rao PS, Lipow HW (1972) Growth retardation in steroid dependent asthma - Management by corticotrophin (ACTH). Clin Pediat 11: 93-97.
-
Tanner JM, Whitehouse RH, Takaishi ML (1966) Standards from birth to maturity for height, weight, height velocity and weight velocity: British children, 1965 - Part II. Arch Dis Child 41(220): 613-635. PMID: 5927918.
-
Friedman M, Strang LB (1969) The effects of corticosteroid and ACTH therapy on growth and on the hypothalamic-pituitary adrenal axis of children. Scand J Respir Dis Suppl 68: 58-69.
-
Rao PS, Molthan ME (1973) Systemic venous anomalies and partial heterotaxia with normal heart. Amer J Dis Child 125(5): 749-752.
-
Rao PS, Jue KL, Isabel-Jones J, Ruttenberg HD (1973) Ebstein’s malformation of the tricuspid valve with atresia. Differentiation from isolated tricuspid atresia. Amer J Cardiol 32: 1004-1009.
-
Van Praagh R, Alido M, Dungan WT (1971) Anatomic types of tricuspid atresia: clinical and developmental implications (abstract). Circulation 44(2): 115.
-
Rao PS, Silbert DR (1974) Superior vena caval obstruction in total anomalous pulmonary venous connexion. Brit Heart J 36(2): 228-232.
- Ramsay Hunt Syndrome Presenting as Gait Imbalance without Facial Paralysis: A Case Report
- Unveiling Hidden Culprits: An Observational Study of Upper Gastrointestinal Endoscopy Findings in Symptomatic Cholelithiasis Patients
- Assessing Health Care Providers’ Proficiency in International Patient Safety Goals: A Study to Assess the Knowledge &Practice on Patient Safety in a Tertiary Care Teaching Hospital in Gujarat
- Challenges in Diagnosing Child Language Disorders
- Stunting Service Management Model in the South-Central Timor Region, East Nusa Tenggara, Indonesia
- Acute Small Bowel Obstruction Presenting as Gangrenous Jejunal Loop Secondary to Intestinal Endometriosis – A Rare Case Report