Neuroprotective Effects of N-Acetyl-Methoxytryptamine in a Neurotoxic Mice Model of Huntington’s Disease Induced by Quinolinic Acid
Huntington’s Disease (HD) is an autosomal dominant neuropathology associated with severe degeneration in neurons of the basal ganglia. It is characterized by progressive motors symptoms, cognitive deficits and dementia. The genetic mutation induces trinucleotide repeat expansion (TNR) of cytosine, adenine and guanine (CAG). The CAG-repeat is translated into a poly glutamine (polyQ) stretch caused by molding of DNA “loop out” structures and DNA slippage during the replication. This expanded sequence encodes a mutant huntingtin (mHtt), which is associated to neuronal dysfunction, cellular death, ubiquitous molecular and cellular anomalies. Furthermore, there are progressive impairments in motor control, cognitive function and mood. Quinolinic acid (QA) is an excitotoxic compound in humans and animals that has been linked to HD. N-acetyl-methoxytryptamine (MLT) is a hormone that produce anti-oxidant and anti-inflammatory effects on cell damage. The principal aim of this study is to know if QA induces damage in striatal astrocytes and neurons and to evidence how MLT can participate as a neuroprotectant agent against the neurotoxic injury. We inoculate QA intrastriatal in 8 weeks old wild type (WT) female mice as endogenous neurotoxic model of HD. Here, we worked with three different groups of mice, and the striatal injection was administrated in all animals. PBS were inoculated in control mice; the second group was injected only QA; and the last group was pre-treated with MLT 30 min before the QA injection. Immunoblots, oxidative stress biomarkers, histological assay of astrocytes and neurons were evaluated as well as rotarod and open field tests. There was a reduction in proteins levels of BDNF and NeuN in QA mice. Contrary effects in lysates showed NF-κB, IκBα, HTT, GFAP and NRs. The analysis of CAT, SOD, Gpx and GSH showed decreased activity, while MDA displayed increased activation in QA mice. The animals pre-treated with MLT revers the tendency of proteins and antioxidant biomarkers; the HTT perinuclear staining in neurons was reduced. NeuN histochemistry showed a greater number of NeuN + cells; the astrogliosis was attenuated; and locomotor activities improved. Analysis of RT-PCR do no show significant diferences between teatments. This study suggests that MLT can induce a compensatory response to neurotoxic stress againts neuro-glial injury. Thus, increasing MLT levels may be a promising treatment for HD disorder.
Introduction
Huntington’s Disease (HD) is an autosomal dominant neuropathology associated with severe degeneration in neurons of the basal ganglia. It is characterized by progressive motors symptoms, cognitive deficits and dementia. The genetic mutation induces trinucleotide repeat expansion (TNR) of cytosine, adenine and guanine (CAG). The CAG-repeat is translated into a poly glutamine (polyQ) stretch caused by molding of DNA “loop out” structures and DNA slippage during the replication. This expanded sequence encodes a mutant huntingtin (mHtt), which is associated to neuronal dysfunction, cellular death, ubiquitous molecular and cellular anomalies. Furthermore, there are progressive impairments in motor control, cognitive function and mood. Quinolinic acid (QA) is an excitotoxic compound in humans and animals that has been linked to HD. N-acetyl-methoxytryptamine (MLT) is a hormone that produce anti-oxidant and anti- inflammatory effects on cell damage. The principal aim of this study is to know if QA induces damage in striatal astrocytes and neurons and to evidence how MLT can participate as a neuroprotectant agent against the neurotoxic injury. We inoculate QA intrastriatal in 8 weeks old wild type (WT) female mice as endogenous neurotoxic model of HD. Here, we worked with three different groups of mice, and the striatal injection was administrated in all animals. PBS were inoculated in control mice; the second group was injected only QA; and the last group was pre-treated with MLT 30 min before the QA injection. Immunoblots, oxidative stress biomarkers, histological assay of astrocytes and neurons were evaluated as well as rotarod and open field tests. There was a reduction in proteins levels of BDNF and NeuN in QA mice. Contrary effects in lysates showed NF- κB, IκBα, HTT, GFAP and NRs. The analysis of CAT, SOD, Gpx and GSH showed decreased activity, while MDA displayed increased activation in QA mice. The animals pre-treated with MLT revers the tendency of proteins and antioxidant biomarkers; the HTT perinuclear staining in neurons was reduced. NeuN histochemistry showed a greater number of NeuN + cells; the astrogliosis was attenuated; and locomotor activities improved. Analysis of RT-PCR do no show significant diferences between teatments. This study suggests that MLT can induce a compensatory response to neurotoxic stress againts neuro-glial injury. Thus, increasing MLT levels may be a promising treatment for HD disorder.
Materials and Methods
Animals
A total of 198 Female C57B1/6 mice (8 weeks) were used from the animal facility of Pharmaceutic Industries Laboratory (LIFE). The rodents were housed 5 per cage in a controlled environment (12/12-h light/dark cycles); the relative humidity is 45%, and the temperature is 75°F±1°F, free access to standard laboratory mice food and water ad libitum. The animal care for this experimental protocol was in accordance with NIH guidelines for the Care and Use of Laboratory Animals and principles presented in the Guidelines for the Use of Animals in Neuroscience Research by the Society for Neuroscience.
Drugs
Quinolinic acid (QA) was dissolved in phosphate buffered saline (PBS, pH 7.0). Melatonin was dissolved in ethylic alcohol (EtOH) then diluted in ultrapure water (Milli-Q) (0.5% final concentration). Both compounds were purchased from Sigma Chemical Co, USA. All other chemical substances were of analytical grade.
Experimental Design
Mice were randomly divided into three groups (n=10) as follows: 1) isotonic saline solution - 0.9% (control); 200 nmol of QA, (QA group); and 3) 10mg/kg (i.p.) of MLT 30 min before of administration of 200 nmol QA (QA+MLT group), [1]. Animals were anesthetized with ketamine/Xylazine (90/10 mg/kg, i.p), and then they were administrated with saline solution or QA unilaterally in the right striatum area with Hamilton syringe. 15 days after of drugs administration, the mice were perfused intracardially. The stereotaxic coordinates to injections were anteroposterior +0.6 mm, mediolateral −2.1 mm, and dorsoventral −2.2 mm from the bregma based in Paxinos and Watson atlas (2007). Body weight was evaluated every three days. The numbers of the animals used in all groups studied in each experiment is the following: 18 mice were used to histological studies (n=6/group); 30 mice to protein (n=10/group); 30 mice to RNA (n=10/group); 30 mice to oxidative stress analysis (n=10/group) and 90 mice to behavioral test (45 open field test, n=15/group; and 45 to rotarod test, n=15/group).
Fixation
Animals were deeply anaesthetized with ketamine/Xylazine (90/10 mg/kg, i.p) and they were perfused through left ventricle. Initially with PBS and subsequently with a fixative solution containing 4% w/v paraformaldehyde in PBS. Following the brains were removed and kept in cold fixative solution for 90 min then they were washed three times in cold solution of sucrose/PBS (5%) and cryoprotected in 25% of sucrose/PBS. Then brains were rapidly frozen at-70°C for 3 hours and stored at -20°C. Coronal 10-µm-thick brain sections were cut using a microtome (Leica). Sections were cryoprotected by immersion in a solution containing 30% (v/v) ethylene glycol and 20% (v/v) glycerol in PBS at -20°C.
Immunohistochemistry
Brain sections from all groups were simultaneously processed in free-floating state as previously described [2]. All antibodies were diluted in a solution with phosphate-buffered saline (PBS), 1% Triton X-100 and 3% normal goat serum. Development of peroxidase activity was carried out with 0.035% w/v 3,3´ diaminobenzidine plus 2.5% w/v nickel ammonium sulfate and 0.1% v/v H2O2 dissolved in acetate buffer 0.1 M pH 6.0. The appropriate IgG controls were used. For fluorescence, the sections were blocking with 10% goat serum, 0.4% Triton X-100, 0.5% BSA in PBS and incubated with the primary antibody at 4°C ON. The appropriate Alexa-Fluor secondary antibodies were diluted in the same solution. Nuclear counterstaining was done with Hoechst (1 mg/ml). The slides were coversliped with Dapi-Fluoromount-G™ (SouthernBiotech) and post-drying were visualized under fluorescent microscope. The dilutions of the primary antibodies were: Anti-Huntingtin, HTT, MAB2166 1:1000 (Sigma-Aldrich); Glial Fibrillary Acidic Protein, GFAP, 1:1000 (Sigma- Aldrich); Neuronal Marker, NeuN, 1:1000 (Abcam). The morphometric analysis was done as previously described [2].
Protein Extraction
Striatal tissue was thawed, weighed and processed by sonication in Triton-X-lysis buffer containing 1% Triton X-100, 50mM Tris-HCl, pH 7.6, 150mM NaCl, 2mM EDTA, 1mM DTT, phosphatase inhibitor cocktail 2, phosphatase inhibitor cocktail 3 and completeTM EDTA-free protease inhibitor cocktail tablet from Sigma as the ratio of 1:10 (1mg tissue = 10ul Triton-X-lysis buffer). The resulting total lysates were assayed for protein concentration by the Pierce BCA method [3].
Immunoblot
For western blot detection samples from total lysates were loaded in 10% Tris-glycine SDS PAGE. The resolved proteins were transferred to Hybond-P membrane (Amersham) and then the membranes were blocked for 1 h in TBS, 0.1% Tween 20 and 5% non-fat dry milk, followed by overnight incubation with primary antibody diluted in the same buffer. For specific proteins, membranes were blocked in TBS, 0.1% Tween 20 and 5% Bovine serum albumin (BSA) and overnight incubation in primary antibody diluted in same buffer. After washing with 0.1% Tween in Tris-buffered saline, the membrane was incubated with peroxidase-conjugated secondary antibody (antibody to Rabbit and Mouse IgG from KPL and Goat IgG from SeraCare (Life Sciences Inc.) for 1h, and then washed and developed using the ECL chemiluminescent detection system (Supersignal® west pico from Thermo Scientific) in Syngene G Box using ichemiXT GENEsys program. Densitometric analyses were performed using Adobe Photoshop CC 2018 and normalized against the signal obtained by reproving the membranes with beta actin. The dilutions for primary antibodies: Anti-Huntingtin, HTT, MAB2166 1:1000 (Sigma-Aldrich); NF-kB/p65, 1:1000 (cell signaling); IκBα, (Cell Signaling); Brain-derived neurotrophic factor, BDNF, 1:500 (Santa Cruz); Glial Fibrillary Acidic Protein, GFAP, 1:1000 (Sigma-Aldrich); Neuronal Marker, NeuN, 1:1000 (abcam); Beta actin 1:10.000 (Cell Signaling); Anti glutamate receptor NMDAR1, NR1, (1:2000, Sigma- Aldrich); NMDAR-2A, and NMDAR-2B (Millipore), anti- brain-derived neurotrophic factor (BDNF) 1:1000 (abcam).
Real time PCR
RNA extraction from mouse brain according to manufacturer's guidelines (Invitrogen Trizol plus purification kit) was performed. RNA quality was assessed with NanoDrop 2000/2000c Spectrophotometer (Thermo Scientific). 2μg RNA was reverse transcribed using OligoDT primers and Reverse transcription system kit (Promega) according to the manufacturer's instructions. Real time PCRs were performed in a 20μl reaction with 1X LightCycler480 SYBR Green I Master (Roche) containing 2μl of cDNA and 20pmol of each specific primer in a LightCycler480 Real-time PCR System (Roche). Minus reverse transcriptase controls were included in each assay. The specific primers analyzed were: mHdh-Fw: 5′-CAGATGTCAGAATGGTGGCT-3′, mHdh Rev: 5′-GCCTTGGAAGAT TAGAATCCA-3′; HTT Fw 5′-TCGTACCACCATTTGTTTTTCA-3, HTT Rev 5- CCTTTGGCCTATGAGATCTGGATGTG-3; NFkB Fw5′- GAAATTCCTGATCCAGACAAAAAC-3′, NFkB Rev 5′- ATCACTTCAATGGCCTCTGTGTAG-3′; IκBα Fw 5′- GCAATCATCCACGAAGAGAAG CC-3′ IκBα Rev 5′- TTACCCTGTTGACATCAGCCCC -3′; BDNF Fw: 5’- AAGTCTGCATTACA TTCCTCGA-3’, BDNF Rev: 5’- GTTTTCTGAAAGAGGGACAGTTTAT -3’; GRIN1 Fw 5′- CGTTCTTGCCGTTGATGA-3′, GRIN1 Rev 5′- GTAAGAGCCAGCAACGGAG-3′; GRIN2A Fw 5′- GGTTTTAAGATTTGTGCCAGG-3′, GRIN2A Rev 5′- CTTAGACCGAGTTGGCAACA-3′; GRIN2B Fw 5′- AGACTATTCGCTTCATGC-3′, GRIN2B Rev 5′- GTGTGTTGTTCATGGCTG-3′; GRIN2C Fw 5′- GAAGGAAAGACACAGGATGCT-3′, GRIN2C Rev 5′- CCCAAATGTCTTCCCAGGT-3′; GFAP Fw 5’- AGAAAGGTTGAATCGCTGGA-3’, GFAP Rev 5’- CGGCGATAGTCGTTAGCTTC-3’; NeuN Fw, 5′- CCAAGCGGCTACACGTCT-3′, NeuN Rv-5′- GCTCGGTCAGCATCTGAG-3′; β-actin Fw: 5’ CACTTTCTACAATGAGCTGCG -3’, β-actin Rev: 5’ CTGGATGGCTACGTACATGG -3’.
Analysis of Oxidative Stress
Levels of oxidative stress from striatum tissues were evaluated using commercial assays kits according the manufacturer’s instructions. The biomarkers used were: Catalase (CAT), Superoxide dismutase (SOD) Sigma Aldrich (Saint Louis, MO, USA), Lipid Peroxidation (MDA) Abcam (Cambridge, MA, USA), glutathione (GSH), glutathione peroxidase (GS-px) Life Technologies (Grand Island, NY).
Behavioral Test
Rotarod analysis was performed using rotarod apparatus at constant speed of 12 rpm over a 120 seconds period [4]. The locomotor activity was measured for 10 min using an activity chamber (Coulbourn Instruments, Allentown, PA, USA) equipped with 16 x 16 horizontal sensors as described Lee et al., 1992. Latency to falling was recorded on three trials, for a maximum of 2 min in each trial. The animals were trained for 2 days before the age of 8 weeks to allow them to become acquainted with the rotarod apparatus. Open field test (OFT) was analyzed one hour after the beginning of the mice dark cycle. One mouse from each group was tested at the same time in different open field boxes (50x50 cm). Over a period of 15 minutes. The total distance traveled was measured. After each trial, the chambers were washed (70% ethanol+12 min to dissipate its smell) to minimize any odors left by mice on previous trials. The mice were tested three times per day during the experimental design.
Statistics Analysis
All experiments and measurement were done 3-4 times showing identical results. Data are represented as mean ± SEM was performed using one-way analysis of variance followed by Bonferroni’s post host tests (GraphPad Software Inc., San Diego, CA, USA), p <0.05.
Results
MLT Decrease Protein Levels of HTT in QA Mice
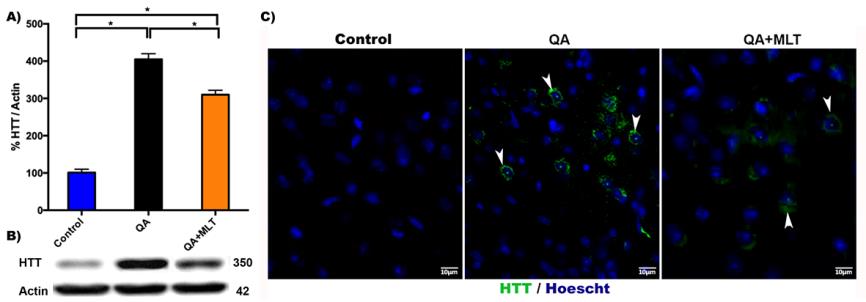
Huntingtin is a protein with approximately 350 kDa of molecular mass. It contains a polyQ near N-terminus, and when trinucleotide CAG expansion on chromosome 4 is more than 37 glutamines, it induces aggregates and accumulates in neuronal nucleus and cytoplasm, most notably in the cerebral striatum. MLT is an endogenous biomolecule with strong broad-spectrum antioxidant properties, which scavenge free radicals and NO in physiological conditions. It has low toxicity, amphiphilic properties and a widespread distribution in the body. The protective effects of MLT play different roles in several pathological conditions including HD. To know if MLT induces a defense mechanism against a neurotoxic model of HD, we first evaluated the HTT protein. There was a significant increase in HTT protein levels from QA mice (Figure 1A). Opposing results show the QA group that was pre-treated with MLT (Figure 1A). This tendency was further visualized by HTT immunofluorescence, which revealed minor perinuclear HTT staining in neurons from the striatal area. (Figure 1B).
Moderate Variations in Genes of the MLT Pre- treated QA Mice
After to demonstrated that MLT reduces levels of the HTT protein in QA mice, we next evaluated if MLT has a regulatory effect on the expression of several genes related to HD. RT-PCR was performed using oligo dT, random primers, and the primers specifically detailed in the methods section. The housekeeping gene (Actin) was also amplified in the same reaction as an internal control. The data analysis indicates mild changes in mRNA relative gene expression between groups analyzed (Table 1).
| Control | AQ | AQ-MLT | |
|---|---|---|---|
| HTT | ND | 0.80±0.3 | 0.06±0.3 |
| mHtt | ND | 0.90±0.1 | 0.05±0.1 |
| NF-kB | 1.18±0.1 | 1.45±0.1 | 1.27±0.1 |
| IKKB | 1.25±0.2 | 1.50±0.2 | 1.38±0.2 |
| BDNF | 1.10±0.2 | 1.25±0.2 | 1.15±0.2 |
| GRIN1 | 1.00±0.1 | 1.09±0.1 | 1.07±0.1 |
| GRIN2A | 1.00±0.1 | 1.08±0.1 | 1.06±0.1 |
| GRIN2B | 1.00±0.2 | 1.11±0.2 | 1.08±0.2 |
| GRIN2C | 1.00±0.1 | 1.07±0.1 | 1.06±0.1 |
| GFAP | 1.00±0.3 | 1.18±0.3 | 1.13±0.3 |
| NeuN | 0.90±0.4 | 1.10±0.4 | 1.00±0.4 |
Table 1: Modest variation in Huntingtin related genes. Relative expression of mRNA levels in striatum. All data are represented a
Attenuation of Oxidative Stress is Induced by MLT in Neurotoxic the Model of HD in Mice
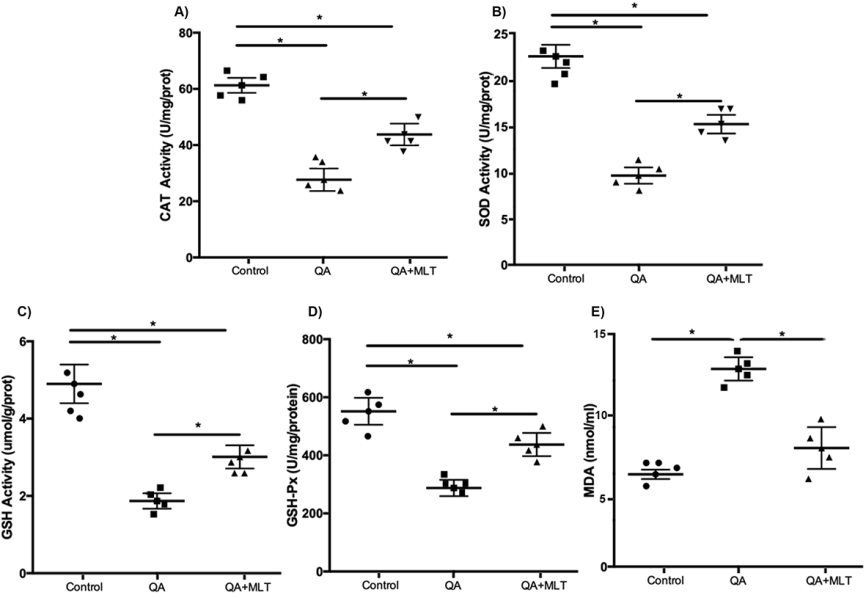
In the inflammatory response, a sustained imbalance between ROS signaling leads to irreversible cellular injury, dysfunction and death. It has been demonstrated that OS is a primary event in HD and that MLT has antioxidant properties against excitotoxicity [5, 6, 7]. Here, we determine the levels of oxidative stress biomarkers in striatal homogenates. Our results show increased levels of SOD and CAT enzymes in QA mice pre-treated with MLT compared to the QA group (Figures 2A-2B). Likewise, our results showed a similar tendency, which showed the quantification of GSH that is the most important scavenger of ROS in a reaction catalyzed by GSH-Px (Figures 2C-2D). The MDA biomarker is an oxidized product to lipid peroxidation in response to oxidative damage in the membrane lipidic. In our experiments, we observed reduced levels of MDA in MLT pre-treated QA mice (Figure 2E).
Decline of Astrodegeneration in MLT Pre- treated QA Mice
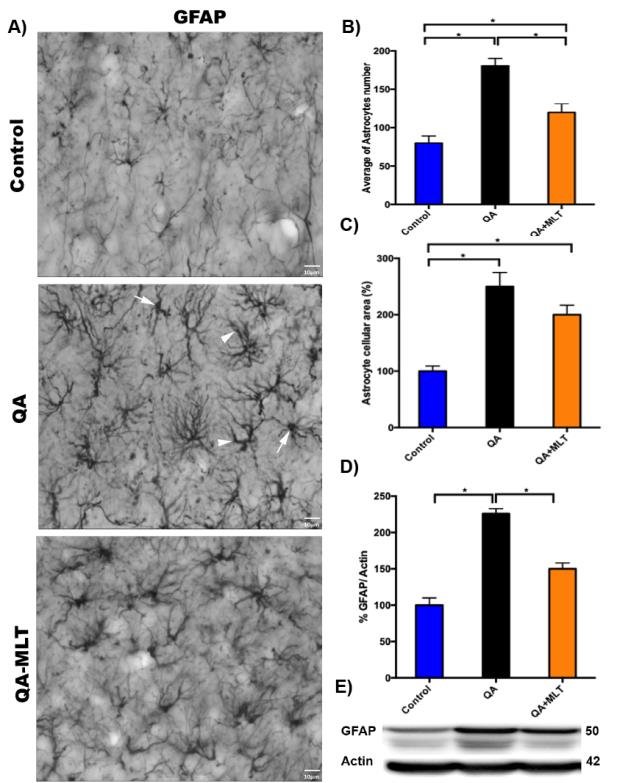
Recently, neuroinflammation has been proposed as the major component of ND. It is well known that astrocytes are specialized cells that provide essential physiological support to neural tissue; they respond to inflammation with molecular, cellular and functional changes. Several similarities between the reactive gliosis in patient and the mice in model of HD have been evidenced in the striatum and cortex from brain tissue [8]. In our experiments, a significant decrease of astrogliosis from striatum was observed by immunostaining of GFAP + cells (Figures 3A). This observation was corroborated analyzing the number (Figures 3B) and cellular area reduced from striatal astrocytes (Figures 3C). Western blot running showed a significant reduction of GFAP protein levels in MLT pre- treated QA mice compared to the QA group and the littermates’ controls (Figures 3D-3E).
NMDA Receptors are Reduced by Induction of MLT in QA Mice
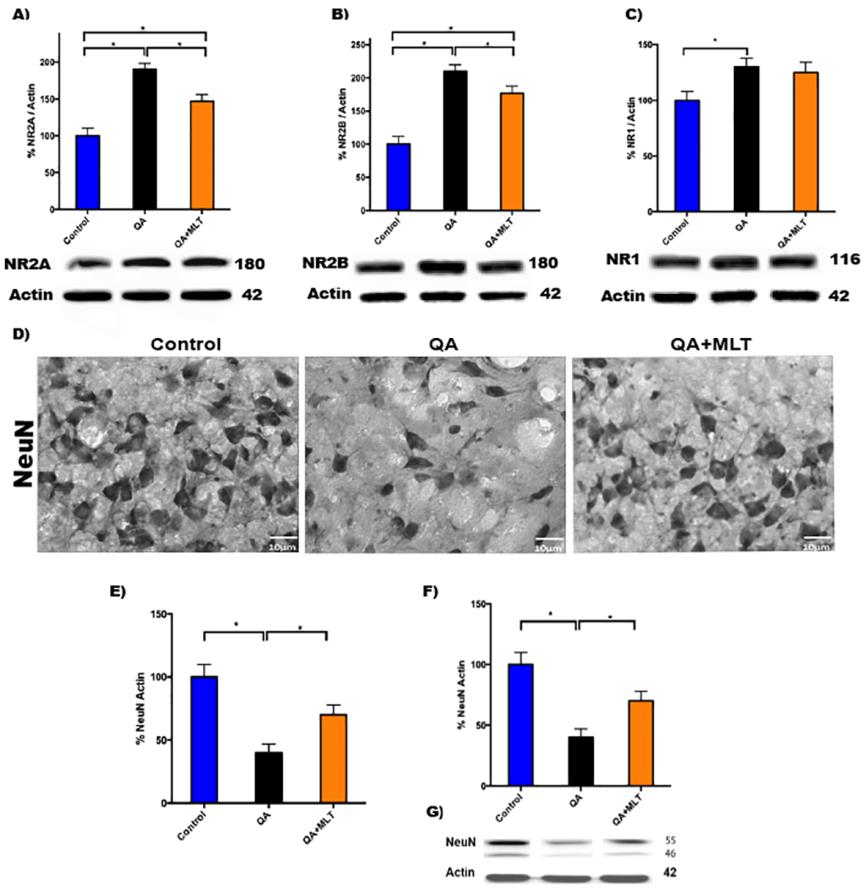
The striatal neurons cells are spiny GABAergic projection neurons and interneurons that have an essential role in planning, modulating the pathway of movements, and other cognitive processes. In HD, the activation of NMDAr mediated by mHtt is a molecular signaling pathway that causes neuronal death. In this study, we report a significant decrease of proteins levels of NR2A and NR2B (Figures 4A-B) in MLT pre-treated QA mice compared with QA mice. NR1 no showed changes (Figure 4C). Then we evaluate the striatal neurons by NeuN immunostaining to correlate NRs data (Figure 4D). The NeuN analysis showed the percentage of neurons/field (Figure 4E), and the western blot finding increased in MLT pre-treated QA (Figures 4F-G).
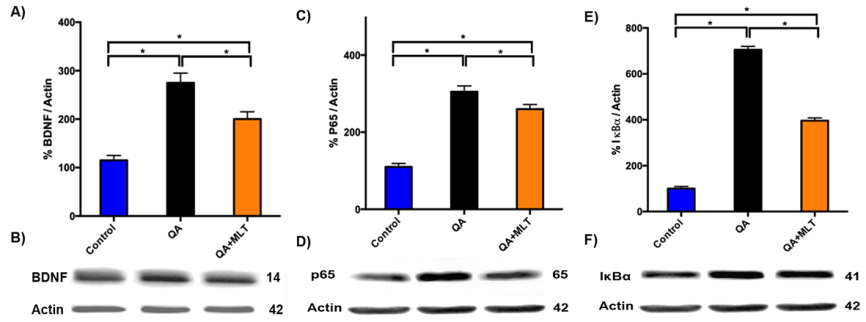
Melatonin Upregulate BDNF to Mediate the Transcriptional Activity of NF-κB in QA Mice
To clarify whether MLT induces changes in the inflammatory NF-κB pathway from QA mice, we analyzed BDNF, because it has been proven that BDNF overexpression plays an important role in HD pathogenesis. To restore striatal BDNF level may have beneficial effects to HD. Immunoblots analysis of BDNF showed reduced proteins levels (Figures 5A-5B), while p65 (Figures 5C-5D) and IκBα (Figure 5E-5F) presented increased expression in QA mice. Contrary effect was shown in the MLT pre-treated QA mice.
MLT Improves Locomotor Activity and Weight in QA Mice
neurotoxic mice model. The Rotarod tests have been widely used as an important indicator of motor coordination. Latency to fall was recorded for five trials over two weeks of assessment, and scores were averaged (Figure 6A).
The locomotor tests were performed to evaluate the effects of the neuromuscular impairments induced by QA
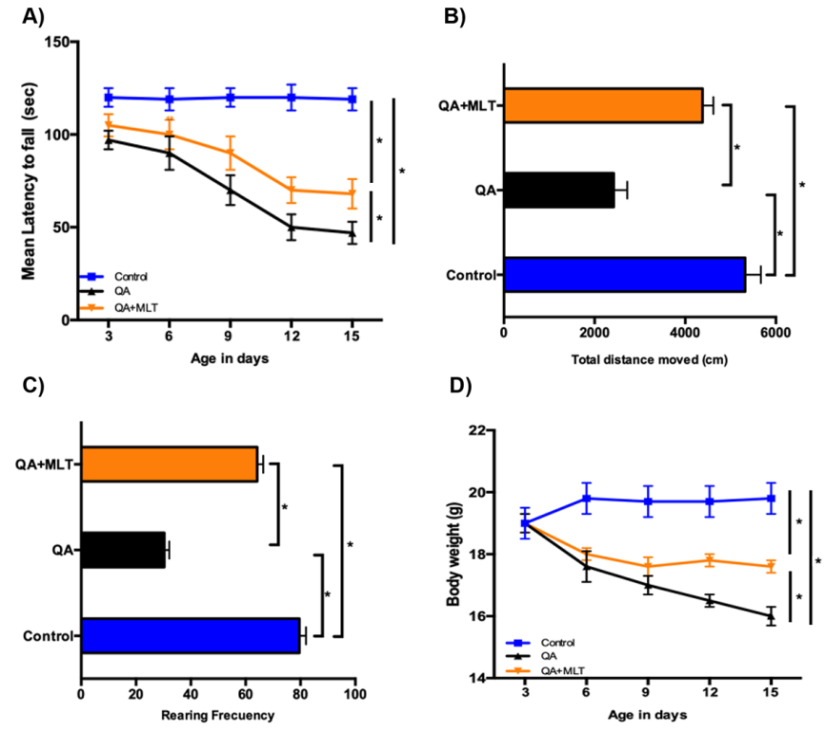
QA mice showed progressive deficits on rotarod with a significantly reduced latency to fall starting at 3 days after the treatment. MLT-treated mice remained longer on the rotarod, which indicates better motor coordination in MLT pre-treated QA mice. In addition, we assessed open field to evaluate the motor function, exploration and general activities. The results showed that QA mice were significantly hypoactive, as measured by the total distance moved (Figure 6B) and rearing frequency (Figure 6C). The MLT pre-treated QA group showed a significant amelioration in OFT results. The total distance covered was higher in the MLT pre-treated QA group than in the only QA mice group (Figure 6C). The QA mice showed a progressive reduction in body weight (Figure 6D); the MLT pre-treated QA group presented increased weight.
Discussion
The induction of the neurotoxic model by QA in the striatum brain is widely used in mice to mimic similar motor disorder and cellular alterations that present patients with HD [9, 10]. In ND, it has been documented that MLT has beneficial functions including reducing OS, accumulating irregular protein and causing cell death [11, 12]. The abnormal expansion of TNR in the HTT gene is caused by mHtt. This change in the DNA sequencing produces buildups of cytoplasmic plaque and neuronal nuclear inclusion in HD [13, 14, 15]. To visualize the endogenous expression of HTT, we used the mAb2166 antibody because it is known to bind strongly to detect the HTT [16]. The QA mice pretreated with MLT showed reduced protein levels of HTT compared with QA mice. Immunofluorescence studies of HTT revealed more perinuclear staining of the N-terminal fragment in striatal neurons from QA mice. These inclusions caused injury and striatal neuronal death. Similar results have been reported in different mice models and human tissue from HD patients [14, 17, 18, 19, 20]. The intranuclear localization of HTT protein occurs early and affects gene transcription in HD. Indeed, microarray studies have displayed a progressive transcriptional change in brain and peripheral tissues [21, 22]. The loss function of transcriptional activator proteins and transcriptional dysregulation has been implicated in the etiology of this neuropathology [23, 24]. The acetylation and deacetylation of histones in nucleosomes is an important process in gene expression. It has been shown in the fruit fly Drosophila model system of HD that the inhibition of histone deacetylase can reverse the reduction of acetylated histones and decrease cellular death [25, 26]. Diversities in samples, protocols and gene detection methods can explain the differences reported in various studies on gene expression. In this study, we found no significant differences in the expression of genes evaluated by RT-PCR. Redox homeostasis is essential for cellular survival. The normal balance between oxidants and antioxidants facilitates ROS and modifies amino acids (AA) of signaling proteins. When oxidants exceed the antioxidant capacity, is leads to a harmful condition of OS. ROS are chemically active in the brain tissue as the excitatory AA and neurotransmitters that are implicated in the pathogenesis of ND. In HD, mHtt have a direct role in mitochondria dysfunction, leading to compromised energy supply and changes in ROS levels [27]. Brain cells have a number of antioxidant molecules and enzymes that restrict the damage and concentrations of ROS, including SOD, CAT, GPXs, GSH [28, 29]. In addition, ROS oxidizes polyunsaturated fatty acids of phospholipid membranes, which causes the formation of oxidized products such as MDA [30]. Studies in serum [31] from HD patients have shown a substantial increase MDA levels, as well as in postmortem HD brain tissue [32]. CAT controls the removal and detoxification of hydrogen peroxide (H2O2), thus decreasing levels of hydroxyl radical formation. Previous reports have demonstrated a very low activity of CAT in HD transgenic mice [33, 34]. SOD activity generates H2O2 and protects cells from the presence of superoxide radical. In Wistars rats, studies have shown that striatal injection of QA induces the reduction of SOD, and there have found a link between neuronal death and SOD activity in the striatum [35, 36]. GSH and Gpx are involved in the oxidative stress response. The function of Gpx is to reduce hydroperoxides levels and their corresponding alcohols with the use of GSH. In the brains of HD patients and R6/1 transgenic mice, the activity of GPx remains unchanged [34, 37]. Our results indicate oxidative striatal brain damage after QA injection, which is evidenced by increased levels of MDA and reduced SOD, CAT, GSH and GPx. This tendency was partially reversed in animals pre- treated with MLT, as a compensatory mechanism that suggests, the antioxidant activity is increased and that MLT induces neuroprotection against enzyme inhibition, as other studies have reported in different neurotoxic models of HD [5, 38, 39]. In the CNS, astrocytes perform homeostatic and regulatory functions to support neuronal cells. The disruption of the normal astrocytic function is the main factor in neurological disorders. In HD brains, a primary response to cellular damage is a prominent astrogliosis, and it increases during the pathology progress [8, 40]. Experiments in vitro show the mHtt expression in astrocytes gets worse as the dysfunction of striatal neurons increases [41]. In vivo, animals expressing mHtt presented similar astrogliosis in the striatum area compared with the brain of HD patients. Here, as mentioned earlier, we obtained analogous astrogliosis findings with increased expressions of GFAP in lysates and immunostaining [17, 42, 43]. Morphological analysis of astrocytes shows the presence of cellular hypertrophy with soma size and cell processes enlarged in QA mice. The excitotoxicity induced by QA causes over activation of NMDAr that increases intracellular Ca2+ [44]; g-aminobutyric acid (GABA) depletion; and ATP consumption and oxidative cell injury [45]. This biochemical alteration in astrocytic cells produces mitochondrial dysfunction, which makes neurons more vulnerable to excitotoxicity, cell injury and death [46]. In our experiments, we found an increased protein expression of NMDAR and a decreased in the number of NeuN+ cells from striatum in QA mice. This evidence is similar in different HD mice models, in concordance with several studies that have revealed striatal neurons were the most vulnerable [47, 48, 49, 50, 51]. However, the effect of MLT in QA mice was to reduce the astrodegeneration that contributes to delaying the neuronal death, as we observed an increased number of NeuN+ cells. The benefit of MLT to stressed cells has been attributed to their ability to scavenge free radicals [52]; moreover, MLT has the capacity to inhibit calcium-induced cytochrome c released from purified mitochondria to keep the necessary energy to maintain physiological functions in the brain [53]. In the inflammatory process, the transcription factor NF-κB plays an important role and BDNF signaling can modulate NF-κB and limit the inflammatory response [54]. Interestingly, in the context of Huntington’s Disease scenery, the mHtt downregulates cortical BDNF transcription, which leads to insufficient support for striatal neurons and causes cellular degeneration [55]. Research has shown decreased BDNF levels in vivo and in vitro models of HD, as well as in the striatum brain and serum of HD patients [56,]. Moreover, a study in astrocytes isolated from R6/2 mice has demonstrated enhanced NF-κB signaling, and others have shown that inducible PC12 cells and striatal cells from R6/2 mice with overexpressed mHtt exon 1 activate NF- κB pathway interacting with IKK [57, 58]. In concordance with those studies, we evidenced an increased protein expression of p65 and IκBa with a reduction of BDNF in QA mice. This tendency was reversed in MLT pre-treated QA mice. We believe that MLT may interact with BDNF to contribute to the anti-inflammatory response by downregulating NF-kB signaling in QA mice. To assess the motor function, body balance and general locomotor activity and willingness to explore, we used the rotarod and open field tests. The impairments induced by QA lesions were ameliorated in mice pre-treated with MLT because they remained on the rotarod longer than QA mice. The general activity and exploration measured by the total distance covered was enhanced as was rearing frequency. These results indicate that MLT improves motor functions in QA mice. On the other hand, the neurotoxic effects of QA in vivo produced progressive alterations in the behavior of QA mice in an exploratory pattern in a new environment, in deprivation of effects on locomotor movement and in spatial and novelty discrimination of the objects. These data are equivalent with other experimental mice, where QA lesion worsened over time, thus mirroring the progressive pathological phenotype presented in HD patients [59, 60, 61]. It is known that weight loss leads to general health weakening in HD patients as well as in mice experiments using different excitotoxins such as kainic acid; quinolinic acid, 3NP or R6/2, and N171-82Q transgenic mice. Here, we demonstrated similar progressive weight loss than other labs that inoculated chemicals compounds or that used transgenics HD mice, as mentioned earlier [62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89]. Interestingly, this weight loss was attenuated in QA mice pre-treated with MLT.
Conclusion
The present study we have shown that QA induces a severe injury in the striatum area, which produces an orchestrated respond of oxidative parameters, variations in proteins, genes and motor dysfunctions in mice. Furthermore, we have evidenced the anti-oxidant and anti-inflammatory effect of MLT on the delay of the disease’s progress, thus enhancing cellular survival from oxidative damage. The potential benefits of MLT may be a possible therapeutic strategy against Huntington’s Disease.
Acknowledgements
This work was support by grants of Pontifical Catholic University, Quito, Ecuador (PUCE); School of Bioanalysis and National Secretary of Science, Technology (SENESCYT) - Prometheus Program, Ecuador awared to RXAR which is Research Associate in University of Wisconsin Madison, USA, SA and PP are Professors from Bioanalysis School-PUCE. MV is researcher from National Health Minister of Peru. We thank McKenna Kohlenberg for the revision of the manuscript, Writing Center Instructor, University of Wisconsin-Madison, USA.
References
-
Williams AJ, Paulson HL (2008) Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci 31(10): 521-528.
-
Aviles-Reyes RX, Angelo MF, Villarreal A, Rios H, Lazarowski A, et al. (2010) Intermittent hypoxia during sleep induces reactive gliosis and limited neuronal death in rats: implications for sleep apnea. Journal of Neurochemistry 112(4): 854-869.
-
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248-254.
-
Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, et al. (1999) Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci 19(8): 3248-3257.
-
Melchiorri D, Reiter RJ, Chen LD, Sewerynek E, Nisticò G (1996) Melatonin affords protection against kainate-induced in vitro lipid peroxidation in brain. Eur J Pharmacol 305(1-3): 239-242.
-
Melchiorri D, Reiter RJ, Sewerynek E, Chen LD, Nisticó G (1995) Melatonin reduces kainate-induced lipid peroxidation in homogenates of different brain regions. FASEB J 9(12): 1205-1210.
-
Tan DX, Reiter RJ, Manchester LC, Yan MT, El-Sawi M, et al. (2002) Chemical and physical properties and potential mechanisms: melatonin as a broad spectrum antioxidant and free radical scavenger. Curr Top Med Chem 2(2): 181-197.
-
Faideau M, Kim J, Cormier K, Gilmore R, Welch M, et al. (2010) In vivo expression of polyglutamine- expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington's disease subjects. Hum Mol Genet 19(15): 3053-3067.
-
Bruyn RP, Stoof JC (1990) The quinolinic acid hypothesis in Huntington's chorea. J Neurol Sci 95(1): 29-38.
-
Rossato JI, Zeni G, Mello CF, Rubin MA, Rocha JB (2002) Ebselen blocks the quinolinic acid-induced production of thiobarbituric acid reactive species but does not prevent the behavioral alterations produced by intra-striatal quinolinic acid administration in the rat. Neurosci Lett 318(3): 137-140.
-
Hardeland R (2005) Antioxidative protection by melatonin: multiplicity of mechanisms from radical detoxification to radical avoidance. Endocrine 27(2): 119-130.
-
Pandi-Perumal SR, BaHammam AS, Brown GM, Spence DW, Bharti VK, et al. (2013) Melatonin antioxidative defense: therapeutical implications for aging and neurodegenerative processes. Neurotox Res 23(3): 267-300.
-
Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, et al. (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90(3): 537- 548.
-
Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, et al. (1999) A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron 23(1): 181-192.
-
Mochel F, Haller RG (2011) Energy deficit in Huntington disease: why it matters. J Clin Invest 121(2): 493-499.
-
Wheeler VC, White JK, Gutekunst CA, Vrbanac V, Weaver M (2000) Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum Mol Genet 9(4): 503- 513.
-
Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, et al. (2001) Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum Mol Genet 10(2): 137-144.
-
Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, et al. (1999) Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N- terminal fragment of huntingtin. Hum Mol Genet 8(3): 397-407.
-
Yamamoto A, Lucas JJ, Hen R (2000) Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell 101(1): 57-66.
-
Ho LW, Carmichael J, Swartz J, Wyttenbach A, Rankin J, et al. (2001) The molecular biology of Huntington's disease. Psychol Med 31(1): 3-14.
-
Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, et al. (2000) Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Hum Mol Genet 9(9): 1259- 1271.
-
Luthi-Carter R, Strand AD, Hanson SA, Kooperberg C, Schilling G, et al. (2002) Polyglutamine and transcription: gene expression changes shared by DRPLA and Huntington's disease mouse models reveal context-independent effects. Hum Mol Genet 11(17): 1927-1937.
-
Harjes P, Wanker EE (2003) The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci 28(8): 425-433.
-
Sugars KL, Rubinsztein DC (2003) Transcriptional abnormalities in Huntington disease. Trends Genet 19(5): 233-238.
-
Borovecki F, Lovrecic L, Zhou J, Jeong H, Then F, et al. (2005) Genome-wide expression profiling of human blood reveals biomarkers for Huntington's disease. Proc Natl Acad Sci USA 102(31): 11023-11028.
-
Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, et al. (2001) Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 413(6857): 739-743.
-
Ross CA, Tabrizi SJ (2011) Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10(1): 83-98.
-
Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O (2012) Oxidative stress and antioxidant defense. World Allergy Organ J 5(1): 9-19.
-
Pisoschi AM, Pop A (2015) The role of antioxidants in the chemistry of oxidative stress: A review. Eur J Med Chem 97: 55-74.
-
Higdon A, Diers AR, Oh JY, Landar A, Darley-Usmar VM (2012) Cell signalling by reactive lipid species: new concepts and molecular mechanisms. Biochem J 442(3): 453-464.
-
Peña-Sánchez M, Riverón-Forment G, Zaldívar- Vaillant T, Soto-Lavastida, Borrero-Sánchez J, et al. (2015) Association of status redox with demographic, clinical and imaging parameters in patients with Huntington's disease. Clin Biochem 48(18): 1258- 1263.
-
Browne SE, Ferrante RJ, Beal MF (1999) Oxidative stress in Huntington's disease. Brain Pathol 9(1): 147-163.
-
Klivenyi P, Andreassen OA, Ferrante RJ, Dedeoglu A, Mueller G, et al. (2000) Mice deficient in cellular glutathione peroxidase show increased vulnerability to malonate, 3-nitropropionic acid, and 1-methyl-4- phenyl-1,2,5,6-tetrahydropyridine. J Neurosci 20(1): 1-7.
-
Pérez-Severiano F, Santamaría A, Pedraza-Chaverri J, Medina-Campos ON, Ríos C, et al. (2004) Increased formation of reactive oxygen species, but no changes in glutathione peroxidase activity, in striata of mice transgenic for the Huntington's disease mutation. Neurochem Res 29(4): 729-733.
-
Rodríguez-Martínez E, Camacho A, Maldonado PD, Pedraza-Chaverrí J, Santamaría D, et al. (2000) Effect of quinolinic acid on endogenous antioxidants in rat corpus striatum. Brain Res 858(2): 436-439.
-
Santamaría A, Pérez-Severiano F, Rodríguez-Martínez E, Maldonado PD, Pedraza-Chaverri J, et al. (2001) Comparative analysis of superoxide dismutase activity between acute pharmacological models and a transgenic mouse model of Huntington's disease. Neurochemical Research 26(4): 419-424.
-
Kish SJ, Morito CL, Hornykiewicz O (1986) Brain glutathione peroxidase in neurodegenerative disorders. Neurochem Pathol 4(1): 23-28.
-
Giusti P, Lipartiti M, Gusella M, Floreani M, Manev H (1997) In vitro and in vivo protective effects of melatonin against glutamate oxidative stress and neurotoxicity. Ann N Y Acad Sci 825: 79-84.
-
Túnez I, Montilla P, Del Carmen Muñoz M, Feijóo M, Salcedo M (2004) Protective effect of melatonin on 3- nitropropionic acid-induced oxidative stress in synaptosomes in an animal model of Huntington's disease. J Pineal Res 37(4): 252-256.
-
Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, et al. (1985) Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 44(6): 559-577.
-
Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, et al. (2005) Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol 171(6): 1001-1012.
-
Gu X, André VM, Cepeda C, Li SH, Li XJ, et al. (2007) Pathological cell-cell interactions are necessary for striatal pathogenesis in a conditional mouse model of Huntington's disease. Mol Neurodegener 2: 8.
-
Gu X, Li C, Wei W, Lo V, Gong S, et al. (2005) Pathological cell-cell interactions elicited by a neuropathogenic form of mutant huntingtin contribute to cortical pathogenesis in HD mice. Neuron 46(3): 433-444.
-
Schwarcz R, Foster AC, French ED, Whetsell WO, Köhler C (1984) Excitotoxic models for neurodegenerative disorders. Life Sci 35(1): 19-32.
-
Foster AC, Whetsell WO, Bird ED, Schwarcz R (1985) Quinolinic acid phosphoribosyltransferase in human and rat brain: activity in Huntington's disease and in quinolinate-lesioned rat striatum. Brain Res 336(2): 207-214.
-
Bano D, Zanetti F, Mende Y, Nicotera P (2011) Neurodegenerative processes in Huntington's disease. Cell Death Dis 2: e228.
-
Feng Q, Ma Y, Mu S, Wu J, Chen S, et al. (2014) Specific Reactions of Different Striatal Neuron Types in Morphology Induced by Quinolinic Acid in Rats. Plos One 9(3): e91512.
-
Guyot MC, Hantraye P, Dolan R, Palfi S, Maziére, et al. (1997) Quantifiable bradykinesia, gait abnormalities and Huntington's disease-like striatal lesions in rats chronically treated with 3-nitropropionic acid. Neuroscience 79(1): 45-56.
-
Han I, You Y, Kordower JH, Brady ST, Morfini GA (2010) Differential vulnerability of neurons in Huntington's disease: the role of cell type-specific features. Journal of Neurochemistry 113(5): 1073- 1091.
-
Ngai LY, Herbert J (2005) Glucocorticoid enhances the neurotoxic actions of quinolinic acid in the striatum in a cell-specific manner. J Neuroendocrinol 17(7): 424-434.
-
Shear DA, Dong J, Gundy CD, Haik-Creguer KL, Dunbar GL (1998) Comparison of intrastriatal injections of quinolinic acid and 3-nitropropionic acid for use in animal models of Huntington's disease. Prog Neuro- Psychoph 22(7): 1217-1240.
-
Lena PJ, Subramanian P (2003) Evaluation of the antiperoxidative effects of melatonin in ammonium acetate-treated Wistar rats. Pol J Pharmacol 55(6): 1031-1036.
-
Wang X, Zhu S, Pei Z, Drozda M, Stavrovskaya IG, et al. (2008) Inhibitors of cytochrome c release with therapeutic potential for Huntington's disease. J Neurosci 28(38): 9473-9485.
-
Xu D, Lian D, Wu J, Liu Y, Zhu M (2017) Brain-derived neurotrophic factor reduces inflammation and hippocampal apoptosis in experimental Streptococcus pneumoniae meningitis. J Neuroinflamm 14(1): 156.
-
Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, et al. (2001) Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science 293(5529): 493-498.
-
Zuccato C, Cattaneo E (2007) Role of brain-derived neurotrophic factor in Huntington's disease. Prog Neurobiol 81(5-6): 294-330.
-
Hsiao HY, Chen YC, Chen HM, Tu PH, Chern Y, et al. (2013) A critical role of astrocyte-mediated nuclear factor-kappaB-dependent inflammation in Huntington's disease. Hum Mol Genet 22(9): 1826- 1842.
-
Khoshnan A, Ko J, Watkin EE, Paige LA, Reinhart PH, et al. (2004) Activation of the IkappaB kinase complex and nuclear factor-kappaB contributes to mutant huntingtin neurotoxicity. J Neurosci 24(37): 7999- 8008.
-
Poucet B (1989) Object exploration, habituation, and response to a spatial change in rats following septal or medial frontal cortical damage. Behav Neurosci 103(5): 1009-1016.
-
Ricceri L, Usiello A, Valanzano A, Calamandrei G, Frick K, et al. (1999) Neonatal 192 IgG-saporin lesions of basal forebrain cholinergic neurons selectively impair response to spatial novelty in adult rats. Behav Neurosci 113(6): 1204-1215.
-
Scattoni ML, Valanzano A, Popoli P, Pezzola A, Reggio R, et al. (2004) Progressive behavioural changes in the spatial open-field in the quinolinic acid rat model of Huntington's disease. Behav Brain Res 152(2): 375-383.
-
Aziz NA, van der Burg JM, Landwehrmeyer GB, Brundin P, Stijnen T, et al. (2008) Weight loss in Huntington disease increases with higher CAG repeat number. Neurology 71(19): 1506-1513.
-
Dunnett SB, Carter RJ, Watts C, Torres EM, Mahal A, et al. (1998) Striatal transplantation in a transgenic mouse model of Huntington's disease. Exp Neurol 154(1): 31-40.
-
Sanberg PR, Calderon SF, Giordano M, Tew JM, Norman AB (1989) The quinolinic acid model of Huntington's disease: locomotor abnormalities. Exp Neurol 105(1): 45-53.
-
Blum D, Galas MC, Pintor A, Brouillet E, Ledent C, et al. (2003) A dual role of adenosine A2A receptors in 3- nitropropionic acid-induced striatal lesions: implications for the neuroprotective potential of A2A antagonists. J Neurosci 23(12): 5361-5369.
-
Chou SY, Lee YC, Chen HM, Chiang MC, Lai HL, et al. (2005) CGS21680 attenuates symptoms of Huntington's disease in a transgenic mouse model. Journal of Neurochemistry 93(2): 310-320.
-
Bolaños JP, Moro MA, Lizasoain I, Almeida (2009) Mitochondria and reactive oxygen and nitrogen species in neurological disorders and stroke: Therapeutic implications. Adv Drug Deliv Rev 61(14): 1299-1315.
-
Bossy-Wetzel E, Petrilli A, Knott AB (2008) Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci 31(12): 609-616.
-
Bradford J, Shin JY, Roberts M, Wang CE, Li XJ, et al. (2009) Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc Natl Acad Sci USA 106(52): 22480- 22485.
-
Bradford J, Shin JY, Roberts M, Wang CE, Sheng G, et al. (2010) Mutant huntingtin in glial cells exacerbates neurological symptoms of Huntington disease mice. J Biol Chem 285(14): 10653-10661.
-
Cepeda C, Ariano MA, Calvert CR, Flores-Hernández J, Chandler SH, et al. (2001) NMDA receptor function in mouse models of Huntington disease. J Neurosci Res 66(4): 525-539.
-
DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, et al. (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277(5334): 1990-1993.
-
Gil-Mohapel J, Brocardo PS, Christie BR (2014) The role of oxidative stress in Huntington's disease: are antioxidants good therapeutic candidates? Curr Drug Targets 15(4): 454-468.
-
Graham RK, Slow EJ, Deng Y, Bissada N, Lu G, et al. (2006) Levels of mutant huntingtin influence the phenotypic severity of Huntington disease in YAC128 mouse models. Neurobiol Dis 21(2): 444-455.
-
Hayden MS, Ghosh S (2012) NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 26(3): 203-234.
-
Khoshnan A, Patterson PH (2011) The role of IkappaB kinase complex in the neurobiology of Huntington's disease. Neurobiol Dis 43(2): 305-311.
-
Kim S, Kim KT (2014) Therapeutic Approaches for Inhibition of Protein Aggregation in Huntington's Disease. Exp Neurobiol 23(1): 36-44.
-
Levine MS, Klapstein GJ, Koppel A, Gruen E, Cepeda C, et al. (1999) Enhanced sensitivity to N-methyl-D- aspartate receptor activation in transgenic and knockin mouse models of Huntington's disease. J Neurosci Res 58(4): 515-532.
-
Liu T, Zhang L, Joo D, Sun SC (2017) NF-kappaB signaling in inflammation. Signal Transduct Target Ther 2.
-
Marini AM, Jiang X, Wu X, Tian F, Zhu D, et al. (2004) Role of brain-derived neurotrophic factor and NF- kappaB in neuronal plasticity and survival: From genes to phenotype. Restor Neurol Neurosci 22(2): 121-130.
-
Mattson MP, Meffert MK (2006) Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ 13(5): 852-860.
-
Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, et al. (2002) Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci 5(8): 731-736.
-
Phatnani H, Maniatis T (2015) Astrocytes in neurodegenerative disease. Cold Spring Harb Perspect Biol 7(6).
-
Reiter RJ, Cabrera J, Sainz RM, Mayo JC, Manchester LC, et al. (1999) Melatonin as a pharmacological agent against neuronal loss in experimental models of Huntington's disease, Alzheimer's disease and parkinsonism. Ann N Y Acad Sci 890: 471-485.
-
Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, et al. (2014) Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol 10(4): 204-216.
-
Soto C (2003) Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci 4(1): 49-60.
-
Takano H, Gusella JF (2002) The predominantly HEAT-like motif structure of huntingtin and its association and coincident nuclear entry with dorsal, an NF-kB/Rel/dorsal family transcription factor. BMC Neurosci 3: 15.
-
Tian F, Hu XZ, Wu X, Jiang H, Pan H, et al. (2009) Dynamic chromatin remodeling events in hippocampal neurons are associated with NMDA receptor-mediated activation of Bdnf gene promoter 1. Amino Acids 109(5): 1375-1388.
-
Zeron MM, Fernandes HB, Krebs C, Shehadeh J, Wellington CL, et al. (2004) Potentiation of NMDA receptor-mediated excitotoxicity linked with intrinsic apoptotic pathway in YAC transgenic mouse model of Huntington's disease. Mol Cell Neurosci 25(3): 469- 479.
- A Review of Gene Therapy for Parkinson's Disease to Control Dopaminergic Neurons
- Late-Onset Myasthenia Gravis in a Patient with Recurrent Breast Cancer: A Case Report
- Covid-Induced Dystonia and Opsoclonus: A Case Report
- Generalized Tonic-Clonic Seizure in a Pediatric Patient with Sunflower Syndrome: A Case Report
- Comparison of Doppler Guided Seldinger Technique Versus Classic Palpatory Seldinger Technique for Radial Artery Cannulation-an Open Label Randomized Controlled Trial
- Brown Sequard Syndrome: Understanding the Complexities of Spinal Cord Injury