A Rare Case of Apert Syndrome due to Mutation of FGFR2 Gene (Fibroblast Growth Factor Receptor 2) – A Disorder of Advanced Paternal Age- A Case Report
The effects of advanced maternal age on offsprings have been well studied and the best documented example is Down syndrome. A small group of congenital disorders, called paternal age effect (PAE) disorders include Apert syndrome, Crouzon syndrome, Achondroplasia and Thanatophoric dysplasia are associated with increased paternal age, relative to the general population. One such case of Apert Syndrome has been discussed in this article.
Introduction
Owing to a recent trend for delaying fatherhood, the genomic integrity of spermatozoa of older men has become a focus of interest. Children of elderly fathers are at a higher risk for certain monogenic conditions collectively termed paternal age effect (PAE) disorders, which include Achondroplasia, Apert Syndrome and Costello Syndrome. These disorders occur due to specific mutations originating exclusively from the male germline, in genes encoding components of the tyrosine kinase receptor/RAS/MAPK signaling pathway [1]. These mutations, occurring randomly during mitotic divisions of spermatogonial stem cells (SSCs), confer a selective growth advantage on the mutant SSC. The clonal expansion of the mutant SSCs generates greater numbers of mutant spermatozoa. This phenomenon, termed selfish spermatogonial selection, is likely to occur in all men.
Case Report
An 8yr. old boy, born of non-consanguineous parents, presented to the genetic clinic of SSKM hospital, Kolkata with abnormally large and misshapen head, dysmorphic face, short stature and malformed hands and feet. His father was of advanced age (45yrs). The antenatal and perinatal periods were uneventful. The malformed hands and feet of the child were noticed at birth. The parents noticed progressive increase in size of their child’s head, which was normal at birth. There was global developmental delay, delayed speech and short stature. On examination, the child had moderate intellectual disability and hypernasal monosyllabic speech. Anthropological measurements revealed macrocephaly, (head circumference was 58cm, >3SD) with short stature (height - 78cm, <3SD) and weight - 13kg (Figure 1). The child had dysmorphic facies with turricephaly (tall, tower shaped skull), brachycephaly (transverse diameter>antero - posterior diameter) and tall, broad forehead, prominent supra orbital ridges, shallow orbits, proptosis, hypertelorism, depressed nasal bridge, high arched cleft palate (Figures 1, 2 & 3).
There was bilateral “mitten hands”- second to fifth digits of both hands showed syndactyli (complete fusion of fingers), with contiguous nail beds (synonychia) and cup shaped palms. The thumbs were proximally placed, broad and radially deviated (hitchhiker’s thumb) (Figure 4). Both feet showed “sock feet” – syndactyli and synonychia of second to fifth toes with broad halluces. There was bilateral genu valgum (Figure 5).
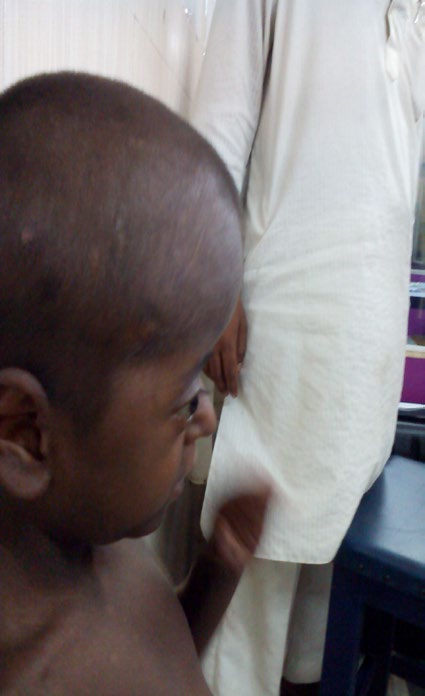
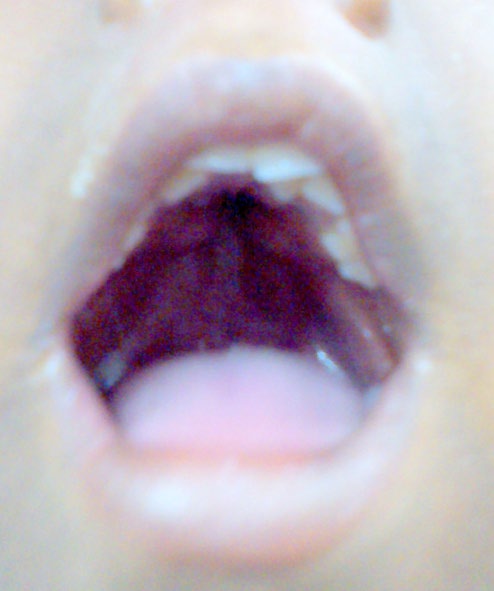
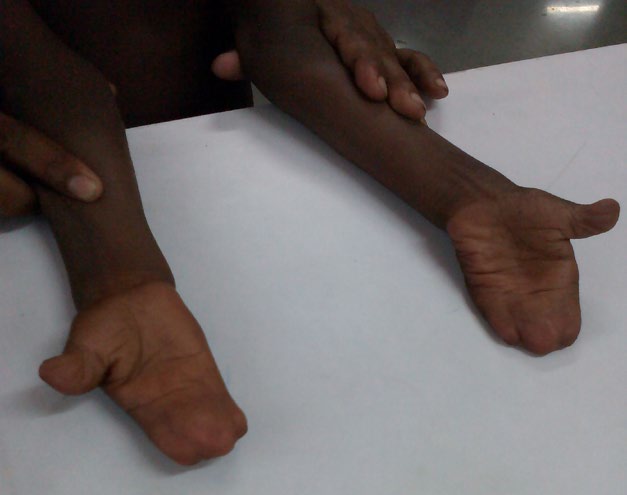
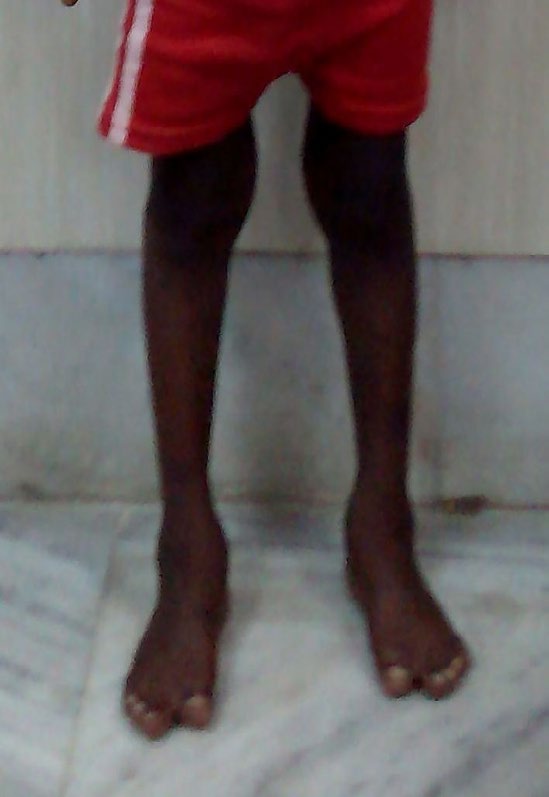
Visual acuity and hearing were normal. Systemic examination revealed no abnormality in cardiovascular, respiratory, gastro intestinal and genitourinary systems.
Investigations
X ray skull revealed bony bridging along coronal sutures (bicoronal synostosis).
Hand and foot radiography – cutaneous and osseos syndactyli; proximal phalanx of thumbs and great toe – delta shaped deformity.
Phenotype and radiological findings suggested the clinical diagnosis as Apert syndrome. Genetic testing was advised for skeletal dysplasia gene panel by next generation sequencing. Report indicated heterogeneous missensense mutation in the FGFR2 gene (c.758C>G p.Pro253Arg) The patient was advised speech therapy following surgical correction of cleft palate.
Management: Cranio-facial surgery, to correct dysmorphic features and orbital surgery to correct proptosis in tertiary centre were advised. Application of bland eye ointment was advised to keep the conjunctiva and cornea moist in view of proptosis.
Genetic Counseling
Apert syndrome is an autosomal dominant disorder, with de novo mutations in most cases. Thus, the risk of recurrence in siblings of proband is minimal (<1%). Usually these patients have decreased reproductive fitness. The de novo mutations are commonly associated with advanced paternal age (Figure 6).

Discussion
Apert syndrome is one of the commonest examples of paternal age effect (PAE) disorders caused by specific missense mutations in the fibroblast growth factor receptor 2 (FGFR2) gene. Other missense mutations in the same gene can give rise to disorders like Crouzon and Pfeiffer syndromes.
Apert Syndrome belongs to the FGFR-related craniosynostosis syndromes. Craniosynostosis is the premature closure of cranial sutures, with a birth incidence of 1 in 3000. In Apert Syndrome, there is bicoronal synostosis in early infancy. The anterior and posterior fontanelles are widely patent. The midline calvarium, with a gaping defect extending from the glabellar area to the posterior fontanelle, permits the accommodation of the growing brain, giving rise to turribrachicephaly, bulging forehead and proptosis. Our patient had similar cranio-facial features.
Unique FGFR2 mutations cause an increased number of precursor cells to enter the osteogenic pathway and lead to increased subperiosteal bone matrix formation and premature calvaria ossification. The molecular basis of the Apert Syndrome is remarkably constant- one of the 2 missense mutations caused by substitution of nucleotides resulting in amino acid substitutions-serine252tryptophan or proline253arginine [2]. Syndactyli is more common with the proline253arginine mutation in the FGFR2 gene [3]. The severe limb abnormalities found in our patient, together with mild cognitive impairment is due to pro253arg mutation in FGFR2 gene.
The paternal age effect (PAE) in Apert Syndrome - PAE disorders are caused by dominant heterozygous mutations, with a triad comprising (1) paternal origin of mutations (2) a advanced paternal age effect and (3) a high germline mutation rate.
Gender Origin of Spontaneous Mutations
Although chromosomal nondisjunctions originate mainly in the female germline, the majority of point mutations and small deletions or insertions tend to be paternal in origin. Spermatogenesis requires regular mitotic divisions of spermatogonial stem cells (SSCs) from puberty onward, at a rate of 23 divisions per year to produce mature sperm cells. This process involves recurrent rounds of DNA replications, so random mutational events may arise in the male germline, explaining the elevated male-to-female mutation ratio between 2 and 7
Paternal Age Effect
The traditional explanation for paternal age effects was age-dependent accumulation of recurrent mutations taking place within localized hypermutable DNA hotspots (the copy- error hypothesis) [4, 5]. In Apert syndrome however, the age excess is very pronounced, rising sharply as the father’s age increases, even after correction for maternal age. Probably other age-dependent factors, such as erroneous DNA replication, inefficient DNA repair mechanisms, or repeated mutagenic exposures may contribute to the accumulation of mutations.
High Germline Mutation Rate
The major determinant of the paternal age effect is expression of mutant protein in Seminiferous Stem Cells (SSC). The mutant SSC gets preferentially selected and undergoes localized clonal expansion. This mechanism was named protein-driven selfish selection [6].
The sperm of all men develop PAE mutations as they age and selfish selection of mutant SSCs is likely to be a universal process.
Conclusion
The sperm of all men show PAE mutations as they age. Prenatal screening via cell - free fetal DNA from maternal blood can be offered to couples with advanced age in the male partner, to screen for PAE disorders like Apert’s syndrome, and achondroplasia [7, 8].
References
-
Maher GJ, Goriely A, Wilkey AO (2014) Cellular evidence for selfish spermatogonial selection in aged human testes. Andrology 2(3): 304-314.
-
Wilkie AOM, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, et al. (1995) Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet 9(2): 165-172.
-
Lajeunie E, Cameron R, El Ghouzzi V, de Parseval N, Journeau P, et al. (1990) Clinical variability in patients with Apert’s syndrome. J Neurosurg 90(3):443-447.
-
Crow JF (2000) The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet 1(1): 40- 47.
-
Penrose LS (1995) Parental age and mutation. Lancet 269(6885): 312-313.
-
Goriely A, McVean GA, van Pelt AM, O’Rourke AW, Wall SA, et al. (2005) Gain- of- function amino acid substitutions drive positive selection of FGFR2 mutations in human spermatogonia. Proc Natl Acad Sci 102(17): 6051-6056.
-
Tang DL, Li Y, Zhou X, Li X, Zheng F (2009) Multiplex fluorescent PCR for noninvasive prenatal detection of fetal-derived paternally inherited diseases using circulatory fetal DNA in maternal plasma. Eur J Obstet Gynecol Reprod Biol 144(1): 35-39.
-
Lim JH, Kim MJ, Kim SY, Kim HO, Song MJ, et al. (2011) Non-invasive prenatal detection of achondroplasia using circulating fetal DNA in maternal plasma. J Assis Reprod Genet 28(2): 167-172.
- Postpartum Maternal Mental Health - A Narrative Review
- Beta HCG in Cervico-Vaginal Secretion as a Predictor of Preterm Delivery
- Successful Management of Mid Trimester Foetal Death with Major Placenta Previa by Expectant Management Followed by Induction of Labour
- To Evaluate the Expression of Egr2 Gene in Term Low Birth Weight Newborns
- Impact of Maternal Obesity on Maternal and Foetal Outcomes: A Prospective Cohort Study from Northern India
- ‘’Benefit of Pulsatile GnRH Therapy in Treatment of Functional Hypothalamic Amenorrhea (FHA) and Congenital Hypogonadotropic Hypogonadism(CHH) in Infertile Patients Over Canonical Gonadotropins with IVF –A Short Communication’’