First Principle Calculations of Phases Stability and Electronic Structure of the Trihalide Perovskite RbSnBr3
The most commonly used and studied halide perovskite is ABX3, where A stands for Rb, B for Sn, and X for Br. A perovskite with high stability and ideal electronic band structure would be of essence, especially considering the materials used for solar cells. In this work, we have considered the two phases (cubic, orthorhombic) of RbSnBr3. the two structures are studied to understand how are the crystal structure stability, band gap and the electronic properties including the total density of states and the projected densities of state have been studied, we found a good agreement with experiments and other calculations. The material has a band gap of 0.57 ev resulting from the transition Br 4P5 → Sn 5P2 that makes the material as a good perovskite compound candidate of a great interest for potential solar-cell applications.
Introduction
Perovskites are a large family of crystalline ceramics that takes their name from a specific mineral called (CaTiO3) because of their crystalline structure. As early as the 1990s, applications of perovskites underwent a major revolution thanks to their low cost for photovoltaic cells. Among the first tests is to introduce perovskite in a photovoltaic cell quadrupled to achieve yields of about 30%, exceeding the yield of silicon cells such as OLEDs (Organic Light Emitting Diodes), transistors and photo detectors [1, 2]. Rubidium-tin III Bromine RbSnBr3 is a perovskite material considered among the class of luminescent materials, it has specific characteristics such First Principle Calculations of Phases Stability and Electronic Structure of the Trihalide Perovskite RbSnBr3 Conceptual paper that: the maximum emission wavelength is about 540 nm [3]. These characteristics are also used to use this material for applications based on the photo-luminous principle such as solar cells. Several studies have been made on luminescent materials [4, 5, 6, 7, 8, 9], the two materials RbSnCl3 and RbSnI3 have been investigated in several studies [10].
Moreover, concerning the material RbSnBr3, our research showed that no work was done except that it was the subject of a single experimental study based on the Raman spectrum measurement as a temperature function between 14 and 520 K [11]. Thanks to its low cost and its ease of transformation, the RbSnBr3 material Phy Sci & Biophy J
is used in several photovoltaic applications such as: solar cells based on DSSC (Dye-Sensitized Solar Cell) architecture [12], optoelectronic devices such as photodetectors [13], light-emitting diodes [14], lasers [15], thin-film transistors [16], and OILEDs [17]. RbSnBr3 is characterized by a photoluminescence of high quantum efficiency, with a narrow spectral width, as well as its very low cost which makes it very attractive for industrialists. This prompts us to further explore this material which is characterized by such remarkable properties and applications in different fields. Our work aims to explore the structural and electronic properties of the three phases (cubic, orthorhombic and triclinic) of this material using the potential Pseudo formalism and the plane wave method "implemented in the Quantum Espresso code [18].
Methodology and Structure
The DFT calculations are performed in the QUANTUM ESPRESSO [14] simulation package with the PerdewBurke-Ernzerhof (PBE-GGA) [15] generalized
gradient approximation (GGA) and plane-waves (PW) pseudopotentials. All calculations are performed with a cut-off of plane waves of 30 Ry and energy cut-off of 120 Ry. The Monkhorst k-point mesh used for these calculations is 6×6×6. The energy convergence of the calculated system is 1 × 10−6 eV.
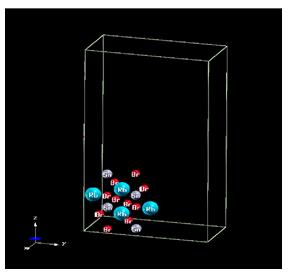
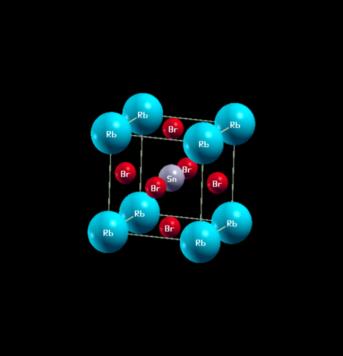
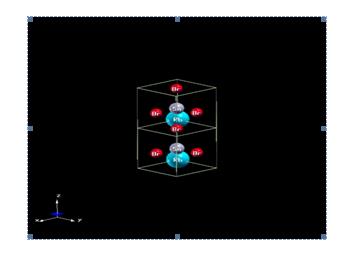
The crystalline structure of RbSnBr3: Among the luminescent materials, there are the conductive materials which have the form, where A is an element of the group 1, such as: rubidium (Rb), B is the element from the group 14 such as tin (Sn), X is the element from the group 17 such as bromine (Br).
Structures
The space group of the cubic structure (Figure 1) of the material RbSnBr3 is Pm3. The orthorhombic structure (Figure 2), is characterized by the space group Pnmn. The triclinic structure (Figure 3), has a space group P1.
Karima Benyahia, et al. First Principle Calculations of Phases Stability and Electronic Structure of the Trihalide Perovskite RbSnBr3. Phy Sci & Biophy J 2019, 3(3): 000123.
Copyright© Karima Benyahia, et al.
Structural Properties
Determining the parameters of the equilibrium structure is the first and fundamental step. In this study, we explore the three phases: cubic, triclinic and orthorhombic of RbSnBr3. The determination of the parameters of the equilibrium lattice, the bulk modulus of compressibility and its derivative are obtained by the extrapolation of the total energy versus the volume E_tot (eV) by the Murnaghan state equation [1]:
𝐵′ [(𝑉0/𝑉)𝐵′
𝐸(𝑉) = 𝐵𝑉
𝐵′ −1 −1] + 𝑐𝑠𝑡 (1)
where 𝑉0, 𝐵 𝑒𝑡 𝐵′ are respectively: the volume of the ground state, the compressibility modulus and its derivative. The equilibrium volume is given by the minimum of the curve E_tot (V):
−1 𝐵′ ⁄
𝑉= 𝑉0 (+ 𝐵′𝑃
(2) ) 𝐵0
The compressibility modulus B is determined by the curvature of this curve:
𝐵= 𝑉𝜕2𝐸
𝜕𝑉2 (3) And its derivative of the compressibility coefficient B 'is determined by fitage with the Murnaghan equation given by the following equation:
𝐵′
𝐸(𝑉) = 𝐸0 + 𝐵0 𝐵′(𝐵′ −1) [𝑉(𝑉0
−𝑉0] + 𝐵0
𝐵′ (𝑉−𝑉0) (4)
𝑉)
Figure 4 shows the curve of variation of total energy as a function of volume of RbSnBr3 for the three phases, from which the equilibrium quantities (V0, a0, B0 and B ') can be obtained by the plane waves PW and the pseudopotential method and which are summarized respectively in the following (Tables 1-3):
| Le matériau | Approximation | a( $A^0$ ) B(Gpa) | B' Total énergie |
|---|---|---|---|
| Cubic | PW, Pseudopotentiel | 5.986 16.03 | 4.27 -1211.3475528 |
| Autre calcul | 5.87 | / |
Table 1: The equilibrium parameter (𝐴°), Total Energy (Ry) and the Bulk Modulus and its derivative (Gpa) for the cubic Table 1: T
| Le matériau | Approximation | A (a.u) B(Gps) | B(a.u) | C(a.u) | B' Total Energie | Total |
|---|---|---|---|---|---|---|
| Orthorhombic | PW, Pseudopotentiel | 8.83 150.78 | 18.01 | 33.91 | 1.002 | -3700.86 |
| Autre calcul | 8.6 | 18.55 | 31.31 | / | / |
Table 2: The equilibrium parameter (𝐴°), Total Energy (Ry) and the Bulk Modulus and its derivative (Gpa) for the Table 2: The equ
| Le matériau | Approximation | A(a.u) C(a.u) | B(a.u) B(Gpa) | B' Total Energie | Total |
|---|---|---|---|---|---|
| RbSnBr3 | PW, Pseudopotentiel | 11.61 37.13 | 11.04 | 12.07 | 5.3 -1213.83 |
| Autre calcul | 11.083 | 11.088 | 11.094 | / |
Table 3: The equilibrium parameter (𝐴°), Total Energy (Ry) and the Bulk Modulus and its derivative (Gpa) for the Table 3: The equ
Karima Benyahia, et al. First Principle Calculations of Phases Stability and Electronic Structure of the Trihalide Perovskite RbSnBr3. Phy Sci & Biophy J 2019, 3(3): 000123.
Copyright© Karima Benyahia, et al.
Band Gap, Total and Partial Density of State
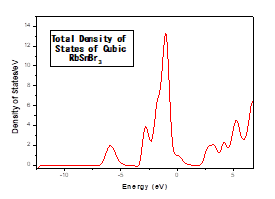
To better understand the electronic properties of the cubic structure of RbSnBr3, it is also interesting and important to determine the total state densities in order to know what kind of hybridizations and which states are responsible in the bonding. To know the nature of the chemical bonds in this material, and the electronic transitions that can take place, we have calculated the total state density (TDOS) which reveals a semiconductor character for this material. The calculated Total State Density (TDOS) is shown in Figure 4.
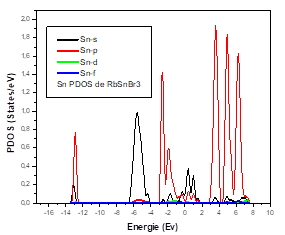
To better understand this total state density (TDOS), we calculated the partial state density of each element constituting the material RbSnBr3.
Karima Benyahia, et al. First Principle Calculations of Phases Stability and Electronic Structure of the Trihalide Perovskite RbSnBr3. Phy Sci & Biophy J 2019, 3(3): 000123.
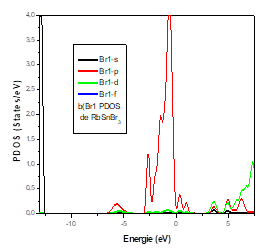
For the Sn atom, we have a strong contribution of the p state in the conduction band and the valence band. For the states, the contribution in the valence band is average. The d State does not contribute in either the valence and the conduction bands.
Copyright© Karima Benyahia, et al.
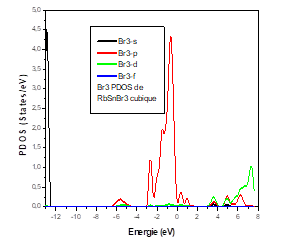
It was found that the top of the valence bands mainly comprised 5p orbital of bromine, while the bottom of the conduction bands is dominated by the 4p orbital of tin.
Conclusion
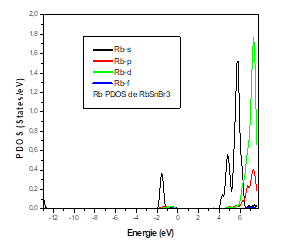
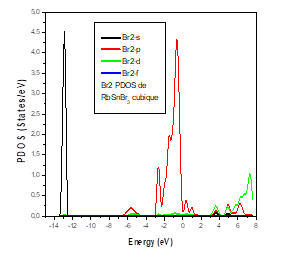
The lattice parameters are in good agreement with those found experimentally and other calculations. The bulk modulus in the cubic and triclinic structures of RbSnBr3 is weak, which shows that the material is not ductile, but we can see that its value is higher in the orthorhombic structure. The total energy reveals that the cubic phase is the most stable structure for RbSnBr3. This material has not been investigated before and our calculations consist of a prediction. The total density of state of RbSnBr3 reveals the semiconductor nature of this material. Moreover, the partial state densities of the different atomic species have shown that the valence band is characterized by a strong presence of the Br-P states, the Sn-P and Rb-s states, d, whereas the conduction band is dominated by the states Sn-P and Rb-s, d.
It was found that the top of the valence bands mainly comprised 5p orbital bromine, while the bottom of the conduction bands is dominated by the 4p orbit of tin. In this material, we found a transition from the electrons of the valence band to the conduction band Br 4P5 → Sn 5P2 corresponding to a small band gap of 0.57 eV which characterize this material as a good photonic absorber and yielded to identify RbSnBr3 as a candidate perovskite compound of a great interest for potential solar-cell applications.
Karima Benyahia, et al. First Principle Calculations of Phases Stability and Electronic Structure of the Trihalide Perovskite RbSnBr3. Phy Sci & Biophy J 2019, 3(3): 000123.
References
-
Im JH, Lee CR, Lee JW, Park SW, Park NG (2011) 6.5% Efficient Perovskite Quantum-Dot-Sensitized Solar Cell_._ Nanoscale 3(10): 4088-4093.
-
Kojima A, Teshima K, Miyasaka T, Shirai Y (2006) Novel Photoelectrochemical Cell with Mesoscopic Electrodes Sensitized by Lead-Halide Compounds (2). 210th ECS Meeting, Cancun, Mexico.
-
Dou L, Yang Y, You J, Hong Z, Chang WH (2014) Solution-Processed hybrid perovskite Phototectors with high detectivity. Nat Commun 5: 5404.
-
Tan ZK, Moghadam RS, Lai ML, Docampo P, Higler R, et al. (2014) Bright light-emitting diodes based on organometal halide perovskite. Nat Nanotechnol 9: 687-692.
-
Deschler F, Price M, Pathak S, Klinberg LE, Jarausch DD (2014) High photoluminescence efficiency and optically pumped lasing in solution-processed mixed halide perovskite semiconductors. J Phys Chem Lett 5(8): 1421-1426.
-
Chin XY, Cortecchia D, Yin J, Bruno A, Soci C (2015) Lead iodile perovskite light-emitting field-effect transistor. Nat Commun 6.
-
Era M, Morimoto S, Tsutsui T, Saito S (1994) Organic- inorganic heterostructure electroluminescent device using a layered perovskite semiconductor (C6H5C2H4NH3)2PbI4. Appl Phys Lett 65(6): 676-678.
-
Chondroudis K, Mitzi DB (1999) Electroluminscence from an organic-inorganic perovskite incorporating a quarterhiophene dye within lead halide perovskite layer_s._ Chem Mater 11(11): 3028-3030.
-
McCarty JG, Wise H (1990) Perovskite catalysts for methane combustion. Catal Today 8(2): 231-248.
-
Gldwasser MR, Rivas ME, Vockic EP (2014) Luminescent materials that emit light in the visible range or the near infrared range and methods of forming thereof. United States Patent.
-
Ketelaar JAA, Rietdijk FAA, Stavern CHV (1937) The crystal structure of ammonium, potassium, rubidium and cesium stannibromide. Rec Trav Chim 56(9): 907-908. Copyright© Karima Benyahia, et al.
-
Kristin P (2016) Materials Data on RbSnBr3 (SG:1) by Materials Project. 36 Materials Science.
-
Le site internet: Brillouni Zone.
-
Giannozzi P, Baroni S, Bonini N, Calandra M, Car R, et al. (2009) QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J Phys: Condens Matter 21(39).
-
Perdew JP, Burke K, Ernzerhof M (1996) Generalized Gradient Approximation Made Simple. Phys Rev Lett 77(18): 3865. Karima Benyahia, et al. First Principle Calculations of Phases Stability and Electronic Structure of the Trihalide Perovskite RbSnBr3. Phy Sci & Biophy J 2019, 3(3): 000123.
-
Monkhorst HJ, Parck DJ (1976) Special points for Brillouin-zone integrations. Phys Rev B 13(12): 5188- 5192.
-
Birch F (1978) Finite strain isotherm and velocities for single‐crystal and polycrystalline NaCl at high pressures and 300°K. J Geophys 83(B3): 1257-1268.
-
Krishnamoorthy T, Ding H, Yan C, Leong W, Baikie T, et al. (2015) Lead-free Germanium Iodide Perovskite Materials for Photovoltaic Application. J Mater Chem A 3(47): 23829-23832. Copyright© Karima Benyahia, et al.
- Sense, Gravity, Parity & Chirality in Mathematical Physics
- Quantum Lattice Simulations PHYSICS: Microcircuit Particle Formation and Observable Macroscopic Irreversible Time - A Discrete Lagrangian with Cellular Automata Framework
- Quantum Biology from Biomacromolecule to Cell, and Central Dogma Described by Quantum Theory
- Focus, Agility, Speed and Technology (FAST) for Sustainability and Growth
- Square Root Metric Geometry and Pati-Salam Model in Curved Space-Time
- A Simple System Demonstrating the Mpemba Effect in Classical Mechanics