Trehalose Stabilizes Lysozyme: A Biophysical and Docking Approach towards Understanding the Mechanism of Co-solute Engineering
Natural selection is believed to be an unforgiving and ceaseless force in the evolution of life on earth. An organism that cannot adopt a changing environment or an environmental hostile to cell functions is at risk as a species. So it is important to understand the mechanism used by the plants, animals and microorganisms in adapting to environments involve such stresses as extremes of temperature, pH, cellular hydration, desiccation, high extracellular salt environments and even the presence of denaturing concentration of urea inside cells. A phylogenetically diverse class of organisms can survive almost complete dehydration. Proteins in vitro lose their structure and function under such stress and, organism should die under such conditions, but organism survives due to presence of some wonder molecules through co-solute engineering by thenature. Included in this list are some plant seeds, fungal spores some nematodes, rotifers and cysts of some crustacean embryos such as the brine shrimp artemia. Nature has adopted a unique method to fight such stress. These organisms typically contain high concentration of many osmolytes that stabilize protein against different stresses and their presence does not alter protein activity. This study shows that trehalose provides the stability to the protein without bringing any change in the protein conformation. The Tm (midpoint of denaturation profile), an index of stability of protein, increases in the presence of trehalose. CD, fluorescence and UV-vis spectroscopy shows that structure of the protein remains intact in a wide range of trehalose. Molecular docking further validated these data.
Introduction
Proteins need to maintain their natively folded structures for proper functions under physiological conditions. However, most proteins are sensitive to changes in cellular and environmental conditions including temperature, pressure, and the presence of salts and other solutes [1, 2]. Therefore, under physiological conditions, proteins require to counter-balance any significant perturbations in these thermodynamic conditions to avoid significant changes in their secondary and tertiary structures [3, 4]. Protein failing to adapt such conditions may result in a partial or complete loss of their functional activity [1]. In order to adapt to such perturbations due to extreme conditions, nature has created certain mechanisms such as accumulation of small organic solutes, also known as ‘‘osmolytes” [3]. Osmolytes are naturally occurring small organic molecules often referred to as ‘‘chemical chaperones”, and are typically accumulated in the intracellular environment at relatively high concentrations that can increase thermodynamic stability of folded proteins without perturbing other cellular processes [5]. A number of cellular osmolytes have been proposed to act as chemical chaperones to rescue defective proteins and to protect native proteins from proteo-toxic intracellular environments. The term “chemical chaperone” was chosen to reflect that though these osmolytes possess properties akin to molecular chaperones, they are not protein molecules that facilitate protein folding and contribute to acquire thermo tolerance. Around 4 decades (1972-2010) have been invested to understand the effect of these compounds on protein structure, folding and function. Therefore, many osmolytes have been selected for their ability to stabilize cell components such as proteins. Osmolytes are small organic solutes accumulated at high concentrations by cells/tissues in response to osmotic stress [2]. Osmolytes increase stability of folded proteins and provide protection against denaturing stresses. In addition to the ability of osmolytes to stabilize proteins, their effects on the functional activity of proteins are equally important, especially at high concentrations. Those having no inhibitory or enhancing effect on the functional activity of enzymes are called compatible osmolytes. It has also been demonstrated that the presence of osmolytes shifts the native conformational equilibrium towards the most compact protein species within native-state ensembles. The mechanism of osmolyte compatibility and osmolyte- induced stability has, therefore, attracted considerable attention in recent years. This approach of nature to stabilize proteins in vivo using cosolutes like glucose, trhalose, glycine, betaine, etc. is called co-solute engineering. Often, these co-solutes or osmolytes are classified as compatible or counteracting based on their effect on the functional activity of proteins. Compatible osmolytes increase protein stability against denaturation with little or no effect on their function under native conditions. Representatives of this class include certain amino acids (e.g., proline and glycine) and polyols (e.g., trehalose, sucrose and sorbitol). Counteracting osmolytes consist of the methylamine class of osmolytes, which are believed to have the special ability to protect intracellular proteins against the inactivating and destabilizing effects of urea and salt [6, 7]. The methylamines includes such as trimethylamine N-oxide (TMAO), glycerol- phosphorylcholine (GPC), glycine betaine (betaine), and sarcosine etc. Counteracting osmolytes stabilize the macromolecule and increase their functional activity, with the additional ability to counteract many natural forces that destabilize proteins. These forces may be perturbing solutes as urea, temperature and hydrostatic pressure [6, 8, 9, 10]. Additionally, there is one more class of osmolyte called Non-compatible osmolytes [11]. This class includes amino acids like, arginine, lysine and histidine. Non-compatible osmolytes perturbs protein stability and enzyme activity as compared to the compatible osmolytes. It is generally believed that these classes of osmolytes are involved in circumventing protein aggregation generated under various proteopathic conditions. Arginine and histidine destabilize RNase A, holo-α-lactalbumin, apo-α-lactalbumin, lysozyme and met myoglobin in terms of the midpoint of the transition curve (Tm) and Gibbs energy change on denaturation (ΔGDo) proteins [11]. The stability of protein refers to the Gibbs free energy released when an unfolded/ denatured polypeptide chain folds back to its stable native conformation. It may also be defined in alternate words as the protein’s resistance to adverse influences such as heat or denaturants that affects the persistence of its molecular integrity or biological function in the face of high temperatures or other deleterious influences. For a reversible folded ↔ unfolded process the stability of a protein is simply the difference in Gibbs free energy, G, between the folded and the unfolded states. The only factors affecting stability are the relative free energies of the folded native (N) (Gɴ) and the unfolded (or denatured, D) (GD) states. The larger and more positive GD, the more stable is the protein to denaturation. A perfectly folded, fully functional monomeric protein can lose its biological activity in vitro by unfolding its tertiary structure to a disordered polypeptide in which key residues are no longer aligned closely enough for continued participation in functional structure stabilizing interactions. Timasheff and co-workers have shown that stabilizing osmolytes are preferentially excluded from the immediate vicinity of the protein domain and this preferential exclusiveness is the main driving force for osmolyte-induced protein stabilization. This preferential exclusion is brought about by three general means. One being the solvophobic interaction between osmolyte and N and D states of proteins; a second means draws from surface tension effect, in that osmolytes that increase surface tension should also be excluded preferentially from the protein surface. A third means of achieving preferential exclusion arises from excluded volume considerations. This volume, called the excluded volume, represents the volume of the solution that is inaccessible to the small molecule [12, 13, 14]. Because of small radius of water molecule than the osmolytes, the excluded volume will be less in case of water molecule, note that because the radius is cubed, a small difference in radius between water and solute can amount to a significant difference between the excluded volume arising from the solute and that arising from water. Only water has access to this difference in excluded volume and this means that for steric (excluded volume) reasons alone, in the presence of solute water will always be enriched in the volume element near the protein surface [13]. Excluded volume has been shown to be the source of the preferential exclusion of PEGs from proteins [15, 16]. Because their radii are larger than that of a water molecule, urea and guanidium chloride are expected to preferentially hydrate proteins for excluded volume reasons. However, preferential interaction measurements on proteins using these solutes show that these strong denaturants do not preferentially hydrate proteins; rather, they favorably interact (bind) with native protein molecules [17, 18].
In fact, misfolding or aggregation occurs frequently [19, 20] inside cells. Polyols such as sucrose, glucose, and sorbitol, are commonly used as protein refolding subordinates to suppress aggregation. There are no significant mechanistic data on how these osmolyte chaperones refold or relieve aggregated/misfolded proteins. One mechanism of protein folding is that it proceeds via hydrophobic collapse wherein a hydrophobic core is first formed followed by secondary and tertiary structural rearrangements to yield the native protein [21]. The hydrophobic core so formed is prone to aggregation because it is largely exposed to the solvent. Polyols, such as sucrose, glucose, glycerol and trehalose are believed to bind to the hydrophobic core, causing a decrease in its hydrophobic character [22]. At the same time, the presence of polyols also increases the viscosity of the refolding buffer, and thus reducing the probability of collision among the hydrophobic core. Recent studies of the products of the sub millisecond folding reactions of several proteins including ribonuclease A [23], lysozyme [24], cytochrome c and apomyoglobin [25] suggest that these proteins fold via accumulation of early intermediates which are structurally highly heterogeneous.
Trehalose is one of the examples of osmolyte that stabilize proteins, raising the free energy of both the native and denatured states, have an unfavorable interaction with the peptide backbone that outcompetes the favorable interaction with the sidechains, which collectively oppose folding.
Improper folding of nascent polypeptides causes many diseases. Such polypeptides are either inactive or have altered activity or are mislocalized due to trafficking defects. Phenylketonuria, the most frequent inborn error of metabolism, is classified as conformational disease. This group includes Alzheimer’s disease, transmissible spongiform encephalitis, serpin deficient disorders, hemolyticanemia, Huntington disease, cystic fibrosis, diabetes type II, amylotropic lateral sclerosis, Parkinson’s disease and more than 15 other less known diseases.
Moreover, organisms accumulate not only one osmolyte but also multiple osmolytes in their biological systems. A few studies have reported the effect of these osmolytes, singly or in combination on the denaturation equilibrium of proteins. The main conclusion of these studies is that all osmolytes plays a protective role for enzyme stabilization by providing an effective approach via alteration of protein microenvironment.
Materials and Methods
Materials and Reagents
Commercial lyophilized preparations of chicken egg- white lysozyme (lot# 061M1328V) was purchased from Sigma Aldrich Co. Hydrochloric acid (HCl), sodium hydroxide (NaOH) pellets, glycine, potassium chloride (KCl), sodium bicarbonate (NaHCO3) and ethylene diamine tetra acetic acid (EDTA) were obtained from Glaxo Laboratories, India and Merck (India) Ltd. respectively. Most of the chemicals and compounds were of analytical grade. Dialysis tubing (molecular weight cut off 6000-8000) was purchased from Spectrum Medical Industries Inc. Millipore filters (pore size 0.22 µm) were bought from Millipore Corporation, and Whatman filter paper no. 1 was obtained from Whatman Laboratories, England. Parafilm used was procurred from American National Co., Chicago. Preparation of Protein and Trehalose Stock Solution Molecular porous membrane having molecular weight cut off in the range 6,000-8,000, which were purchased from Spectrum Medical Industries (USA) were used to dialyse chicken egg-white lysozyme protein solution. Dialysis tubings were essentially prepared according to the procedure recommended by Dr. McPhie as described above (McPhie, 1972). Required amount of lysozyme powder was dissolved in 10 ml of 0.1 M KCl solution. 10 ml of this solution was dialysed against the several changes of 0.1 M KCl solution at pH 7.0 and 4 oC. The protein was then filtered through Millipore filter of pore size 0.22 µm. The stock solution of protein was stored at 4oC. Concentration of the lysozyme was determined experimentally using molar absorption coefficient (M-1cm-1) values of 39000 at 280 nm for lysozyme. The required amount of trehalose was dissolved in the respective buffer. The pH of the solution was maintained to pH 2.0 and filtered through the Whattman filter paper No. 1 and stored at room temperature.
Absorption Spectroscopy
Spectral measurements were made in Jasco V-660 UV/Vis spectrophotometer equipped with a peltier type temperature controller (ETC-505T). The spectra were recorded in the region 340-220 nm. Baseline correction was always carried out. Spectrum of the native protein was recorded against buffer and stored. Spectrum of the protein in the presence of the denaturant was recorded against the same concentration of the denaturant and stored. Difference spectra were obtained by subtracting the spectrum of the native protein from that of the protein in the presence of the denaturant.
Analysis of Thermal Denaturation Curves
Thermal denaturation studies were carried out in Jasco V-660 UV/Vis spectrophotometer equipped with a Peltier
type temperature controller (ETC-505T). Each sample was heated from 20 to 85 °C. Change in optical probe with increasing temperature was recorded. About 650 data points for each transition curves were collected.
After denaturation the sample was immediately cooled down to measure reversibility of the reaction. All blank solutions were subtracted before the data analysis. The absorbance value and CD signal were converted into molar absorption co-efficient ($M^{-1}cm^{-1}$) and mean residue ellipticity ($deg cm^2 dmol^{-1}$), respectively, at a particular wavelength $\lambda$.
Each heat-induced transition curve was analyzed for Tm and $\Delta Hm$ using a non-linear least-squares analysis according to the relation:
$$y(T) = \frac{y_N(T) + y_D(T) \exp[-\Delta H_m / R(1/T - 1/T_m)]}{1 + \exp[-\Delta H_m / R(1/T - 1/T_m)]}$$ (1)
where $y(T)$ is the optical property at temperature $T$ (K), $y_N(T)$ and $y_D(T)$ are the optical properties of the native and denatured protein molecules at temperature $T$ and $R$ is the universal gas constant. In the analysis of the transition curve, it was assumed that a parabolic function describes the dependence of the optical properties of the native and denatured protein molecules i.e., $y_N(T) = a_N + b_NT + c_NT^2$ and $y_D(T) = a_D + b_DT + c_DT^2$, where $a_N, b_N, c_N, a_D, b_D$ are temperature independent coefficients. Value of the constant-pressure heat capacity change ($\Delta C_p$) was determined from slope of the linear plots of $\Delta H_m$ versus $T_m$, using the relation:
$$\Delta C_p = \left( \partial \Delta H_m / \partial T_m \right)_p$$ (2)
Using values of $T_m$, $\Delta H_m$ and $\Delta C_p$, the value of $\Delta G_D$ at any temperature $T$, $\Delta G_D(T)$ was estimated with the help of the Gibbs-Helmholtz equation:
$$\Delta G_D(T) = \Delta H_m \left( \frac{T_m - T}{T_m} \right) - \Delta C_p \left[ (T_m - T) + T \ln \left( \frac{T}{T_m} \right) \right]$$ (3)
Change in Enthalpy and entropy at 25°C was calculated according to these equations:
$$\Delta H_D^0 = \Delta H_m - \Delta C_p (T_m - 298.15)$$ (4)
$$\Delta S_D^0 = \Delta S_m + \Delta C_p \ln (298.15 / T_m)$$ (5)
where $\Delta H_D^0$ and $\Delta S_D^0$ are the change in enthalpy and entropy at 25°C. $\Delta S_m$ is the change in entropy at $T_m$.
Circular Dichroism Spectroscopy
Circular dichroism studies were carried out to characterize secondary structure of the protein. Far-UV CD studies were carried out in Jasco Spectropolarimeter (model J-715) equipped with peltier-type temperature controller (PTC-348 WI) and interfaced with personal computer. Cells of path length cells 0.1 cm was used for far-UV CD. CD instrument was routinely calibrated with d-10 camphorsulphonic acid. Baseline correction was always carried with buffer in question and data stored. The spectrum of the native protein and protein in the trehalose solution were retained and data acquisition was carried out using the J700 software provided by Jasco. The baseline of the buffer solution was subtracted from the native and baseline of trehalose blank from protein with glucose solution respectively. At least 3 accumulations of the scanning were carried to average out the spectrum to improve upon signal to noise ratio in each case including the baseline. $N_2$ was flushed continuously through the machine at the rate of 5 lt/min or higher (below 200 nm) to minimize the noise level. CD data were reduced to concentration independent parameter $[\theta]_\lambda$ (deg cm$^2$ dmol$^{-1}$) which is the mean residue ellipticity using the relation:
$$[\theta]_\lambda = M_0 \theta_\lambda / 10lc$$ (6)
$\theta_\lambda$ is observed ellipticity in millidegrees at wavelength, $M_0$ is the mean residue weight of the protein, $c$ is the protein concentration in gm/cm$^3$, $l$ is the path length of the cell in centimeter.
Fluorescence Spectroscopy
Fluorescence spectra were measured in Jasco FP-6200 spectroflourometer in a 5 mm quartz cell at 25°C, with both excitation and emission slits set at 5 mm. Protein concentration for all the experiments was 0.5 mg/ml. Tryptophan (Trp) residues of the lysozyme were excited at a wavelength of 295 nm and emission spectra were recorded in the wavelength region of 300-400 nm.
Molecular Docking
Various open source tools are utilised for the purpose of analysing and seeing the interactions such as MGL Tools, Auto Dock Tools 4.2, Discovery studio. All the tools were used for finding out the Gibbs Free Energy for binding as measurement of interaction and analysing small molecules trehalose, Trehalose is a disaccharide formed by a 1,1-glycosidic bond between two $\alpha$-glucose units. as low-affinity inhibitors against Lysozyme to validate the result with biophysical results already done. Crystal structures were downloaded from Protein Data Bank. In order to get deep insight into the interaction patterns of trehalose with lysozyme. molecular docking studies were carried out using Auto-Dock 4.2 software. Trehalose is a disaccharide formed by a 1,1-glycosidic bond between two α-glucose units. primarily required input file was prepared by Chemdraw while MM2 force field (Chemdraw versions 12, 12 ChemBio3D) was used for energy minimization The .pdb was converted into pdbqt (ligand and receptor file) i.e. trehalose and myoglobin. entire amino acid residues of the protein were covered with Autogrid 4 module that is blind docking. Herein, the X, Y, Z three dimension of the grid were set to be 51X, 56Y and 47Z Å (coordinates of receptor axes and assigned grid space size as 0.375 Å. Docking were performed using the Lamarckian Genetic Algorithm (LGA) to produce the best conformation of the macromolecule and the ligand. Finally, the docked pose was directly visualized by PyMOL.
Results
Thermal Denaturation Studies of Lysozyme in the Presence of Trehalose
Heat-induced denaturation studies of lysozyme were carried out in the presence of trehalose at pH 2.0 by following changes in molar absorption coefficient (M-1cm-1) at 300 nm as a function of temperature. Figure 1 shows the representative denaturation curves of lysozyme in the absence and presence of trehalose at pH 2.0 inset shows the plot of ΔTm versus the concentration of trehalose. Denaturation at each pH was reversible in the entire range of different concentrations of trehalose. It was observed that the temperature dependence of yN of lysozyme depends on neither the concentration of trehalose nor pH. However, yD depends on pH.
∆Tm, oC Control 0.25 M Trehalose 0.50 M Trehalose 0.75 M Trehalose 1.00 M Trehalose 1.25 M Trehalose 1.50 M Trehalose Figure 1: Thermal denaturation of lysozyme in the absence and presence of different concentrations of trehalose at pH 2.0.
Each transition curves at fixed pH and different concentration of trehalose was analyzed for ΔHm and Tm. Values of ΔHm and Tm are given in Table 1. It is seen in this table that the value of Tm increase with increasing the concentration of trehalose, whereas ΔHm remains insignificantly changed.
| [trehalose], M | Tm, °C | ∆H , ∆S , m m kcal mol-1 kcal mol 1K-1 | ∆H °, T∆S °, D D kcal mol-1 kcal mol-1 | ∆G °, D kcal mol-1 | %∆∆G ° D |
|---|---|---|---|---|---|
| 0 | 57.4 ± 0.1 | 87 ± 4 0.263 | 36 30 | 5.92 ± 0.30 | 0 |
| 0.25 | 59.4 ± 0.2 | 88 ± 2 0.265 | 36 30 | 6.34 ± 0.12 | 7 |
| 0.5 | 61.5 ± 0.2 | 90 ± 3 0.269 | 37 30 | 6.74 ± 0.20 | 14 |
| 0.75 | 63.3 ± 0.3 | 92 ± 2 0.273 | 37 30 | 7.18 ± 0.13 | 21 |
| 1 | 65.6 ± 0.3 | 93 ± 4 0.275 | 37 29 | 7.60 ± 0.18 | 28 |
| 1.25 | 67.4 ± 0.2 | 95 ± 2 0.279 | 38 30 | 8.07 ± 0.13 | 36 |
| 1.5 | 69.1 ± 0.2 | 96 ± 3 0.28 | 38 29 | 8.45 ± 0.21 | 43 |
Table 1: It is seen in this table that the value of Tm increase with increasing the concentration of trehalose, whereas ΔHm remai
Fluorescence Study
To find out the effect of trehalose on the tertiary structure of lysozyme, we have carried out tryptophan fluorescence at pH 2.0 and 25°C in the absence and presence of trehalose.
Excitation wavelength for tryptophan fluorescence was 295 nm and λmax emission was 342 nm for native protein. As we add the trehalose in the native lysozyme there is no significant change in the fluorescence intensity and there is no shift in spectrum peak (Figure 2).
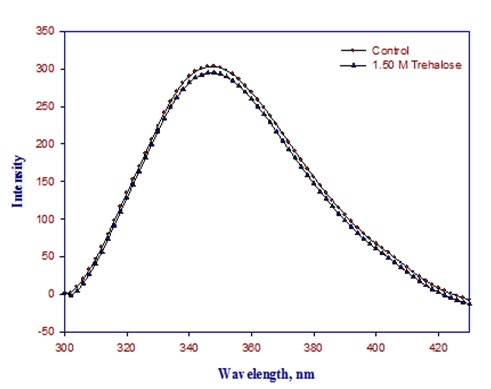
3.3. Far-UV and near-UV circular dichroism spectroscopy of native and denatured lysozyme in the absence and presence of Trehalose.
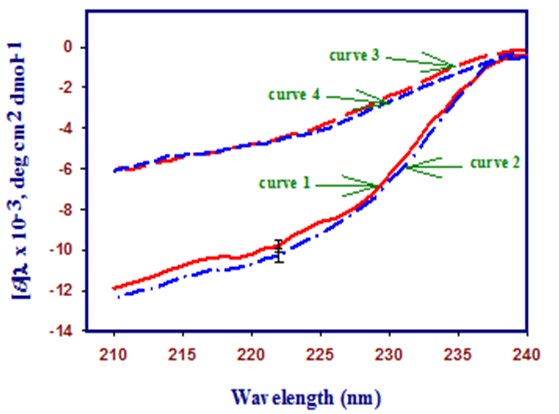
Figure 3 shows the far-UV CD spectra of the native and denatured lysozyme in the absence and presence of highest concentration (1.50 M) of trehalose at pH 2.0. It is seen in this figure that there is no significant change in the secondary structure of the native protein (curve 1 and 2) and denatured protein (curve 3 and 4) upon addition of trehalose.
0.4 [θ]λ x 10-3, deg cm2 dmol-1
260 280 300 320 Figure 4: Near-UV CD spectra of lysozyme in the absence and presence of highest concentration of trehalose (1.50 M). Curve 1 shows the native state of lysozyme at 25°C, curve 2 shows lysozyme with 1.50 M trehalose at 25°C, curve 3 shows lysozyme at 85°C and curve 4 shows lysozyme with 1.50 M trehalose at 85°C.
Figure 4 shows the near-UV CD spectra of the native and denatured lysozyme in the absence and presence of highest concentration (1.50 M) of trehalose at pH 2.0. It is also seen in this figure that there is no significant change in the tertiary structure of the native protein (curve 1 and 2) and denatured protein (curve 3 and 4) upon addition of trehalose.
These Figures (Figures 3 & 4) also show the far- and near- UV spectra of the heat-denatured lysozyme in the absence and presence of highest concentration of trehalose at 85°C (curve 3 and 4). These measurements suggest that there is no significant difference in the characteristic CD spectra of the heat denatured protein in the absence and presence of trehalose.
Molecular Interactions of Lysozyme with Trehalose
In order to get an insight into the interaction patterns of trehalose with lysozyme, molecular docking studies were carried out using AutoDock4 software. Trehalose is a disaccharide formed by a 1,1-glycosidic bond between two α-glucose units. The computational analysis showed that binding interaction exists between trehalose and lysozyme. The value for the binding affinity for trehalose with lysozyme was found to be – 6.4 kcal/mol indicating the favourable interactions of trehalose with lysozyme. The docking studies showed that trehalose interacts with the four conventional hydrogen bonds with Asparagine 59, Aspartic Acid 48, serene 50, Tryptophan 62 via single hydrogen bond (Figure 5).
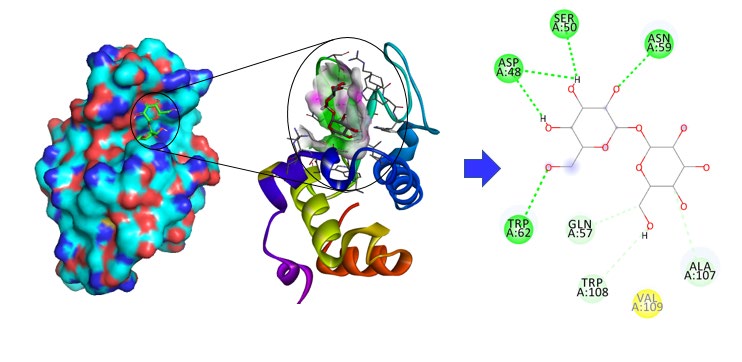
The value for the binding affinity for trehalose with lysozyme was found to be – 6.4 kcal/mol, which corelate with CD and spectroscopy data.
Discussion
To investigate the effect of trehalose on the structure and stability of lysozyme, we have carried out CD, fluorescence and thermal denaturation. It is seen in Table 1 that Tm of lysozyme at pH 2.0 increases linearly with increase in the concentration of trehalose. The stabilizing effect of protein by sugar is explained by the preferential hydration effect or osmophobic theory and volume exclusion effect. Trehalose is preferentially excluded from the protein domain i.e. protein is preferentially hydrated in the presence of trehalose. Preferential exclusion of trehalose from protein domain is more for the denatured state than the native state. Therefore, denature state is more unstable than the native state. Thus the denature equilibrium (N state ↔D state) is shifted toward N (native) state in the presence of trehalose. Stabilizing osmolytes destabilizes the states of protein, but the denatured state is highly destabilizing in comparison to the native one. Thus the protein acquires less destabilized state i.e. native state. We have chosen pH 2.0 for our study because lysozyme is a highly stable protein and at high pH values we do not get complete thermal transition. But at pH 2.0, lysozyme gets denatured and we get complete thermal transition.
The ΔHm values of many proteins remains unchanged in the presence of glucose osmolyte near neutral pH have been reported [26, 27, 28]. This aspect justify the volume exclusion effect of stabilizing osmolyte in which stabilizing osmolytes are completely excluded from protein domain and are not specifically bind to any states (N and D states) of the protein. One of the conclusions of osmolytes studies is that the ΔHm of protein in the presence of osmolytes may increase, decrease or remain unchanged. It is noteworthy that the additives that have relative affinity towards protein results in the increase or decrease of ΔHm of the proteins [29]. In case of destabilizer, the preferentially binding of the protein results in the decrease of ΔHm of proteins [29]. It can be seen in table 1 that the value of ΔHm of lysozyme in the presence of different concentrations of trehalose at all pH values shows insignificant dependence on the concentration of the trehalose. This observation suggests that trehalose has no significant affinity for the lysozyme.
ΔHD o and TΔSD o also show no significant change in the presence of trehalose. This shows that trehalose has no affinity for the lysozyme and completely excluded from the protein domain. Our results justify the preferential exclusion of osmolytes from protein surface. Protein folding in the presence of trehalose is enthalpicaly driven process.
Far-UV and near-UV CD studies show that trehalose does not perturb the secondary and tertiary structure of lysozyme in native as well as in denatured state. It only increases the number of native molecule and decrease the turnover number of protein. Fluorescence spectrum also shows that there is no change in the environment of tryptophan molecule. Thus, trehalose provides the stability to the protein without bring any change in the protein conformation.
Molecular docking shows interactions between protein and osmolyte, however, small value for the binding affinity for trehalose with lysozyme (–6.4 kcal/mol) justify that trehalose do not perturb the secondary or tertiary structure of the protein. We may conclude that trehalose stabilizes the protein during thermal denaturation without affecting the structure of the protein. It is commonly seen that organism which faces heat stress, they accumulate excess of trehalose. Trehalose help them against desiccation, and further increases the thermal stability of the protein to off shoot the impact of high temperature.
References
-
Pace CN (1975) The stability of globular proteins. CRC Crit Rev Biochem 3(1): 1-43.
-
Yancey PH, Clark ME, Hand SC, Bowlus RD, Somero GN (1982) Living with water stress: evolution of osmolyte systems. Science 217(4566): 1214-22.
-
Bolen DW (2001) Protein stabilization by naturally occurring osmolytes. Methods Mol Biol 168: 17-36.
-
Baldwin RL (1999) Protein folding from 1961 to 1982. Nat Struct Biol 6(9): 814-817.
-
Burg MB (1995) Molecular basis of osmotic regulation. Am J Physiol 268(6 Pt 2): 983-996.
-
Lin TY, Timasheff SN (1994) Why do some organisms use a urea-methylamine mixture as osmolyte? Thermodynamic compensation of urea and trimethylamine N-oxide interactions with protein. Biochemistry 33(42): 12695-12701.
-
Yancey PH, Somero GN (1979) Counteraction of urea destabilization of protein structure by methylamine osmoregulatory compounds of elasmobranch fishes. Biochem J 183(2): 317-323.
-
Somero GN (1986) Protons, Osmolytes, and Fitness of Internal Milieu for Protein Function. American Journal of Physiology 251(2): 197-213.
-
Wang A, Bolen DW (1997) A naturally occurring protective system in urea-rich cells: mechanism of osmolyte protection of proteins against urea denaturation. Biochemistry 36(30): 9101-9108.
-
Baskakov I, Wang A, Bolen DW (1998) Trimethylamine- N-oxide counteracts urea effects on rabbit muscle lactate dehydrogenase function: a test of the counteraction hypothesis. Biophys J 74(5): 2666-2673.
-
Rishi V, Anjum F, Ahmad F, Pfeil W (1998) Role of non- compatible osmolytes in the stabilization of proteins during heat stress. Biochem J 329(Pt1): 137-143.
-
Schachman HK, Lauffer MA (1949) The hydration, size and shape of tobacco mosaic virus. J Am Chem Soc 71(2): 536-541.
-
Schellman JA (2003) Protein stability in mixed solvents: a balance of contact interaction and excluded volume. Biophysical Journal 85(1): 108-125.
-
Timasheff SN (1998) Control of protein stability and reactions by weakly interacting cosolvents: The simplicity of the complicated. Advances in Protein Chemistry 51: 355-432.
-
Arakawa T, Timasheff SN (1985) Mechanism of poly (ethylene glycol) interaction with proteins. Biochemistry 24(24): 6756-662.
-
Bhat R, Timasheff SN (1992) Steric exclusion is the principal source of the preferential hydration of proteins in the presence of polyethylene glycols. Protein Sci 1(9): 1133-1143.
-
Arakawa T, Timasheff SN (1984) Protein stabilization and destabilization by guanidinium salts. Biochemistry 23(25): 5924-5929.
-
Timasheff SN (1992) Water as Ligand - Preferential Binding and Exclusion of Denaturants in Protein Unfolding. Biochemistry 31(41): 9857-9864.
-
Speed MA, King J, Wang DI (1997) Polymerization mechanism of polypeptide chain aggregation. Biotechnol Bioeng 54(4): 333-343.
-
Fink AL (1998) Protein aggregation: folding aggregates, inclusion bodies and amyloid. Fold Des 3(1): 9-23.
-
Cheung MS, Garcia AE, Onuchic JN (2002) Protein folding mediated by solvation: water expulsion and formation of the hydrophobic core occur after the structural collapse. Proc Natl Acad Sci U S A 99(2): 685-690.
-
Leandro P, Gomes CM (2008) Protein misfolding in conformational disorders: rescue of folding defects and chemical chaperoning. Mini Rev Med Chem 8(9): 901- 911.
-
Houry WA, Rothwarf DM, Scheraga HA (1996) Circular dichroism evidence for the presence of burst-phase intermediates on the conformational folding pathway of ribonuclease A. Biochemistry 35(31): 10125-10133.
-
Morgan CJ, Miranker A, Dobson CM (1998) Characterization of collapsed states in the early stages of the refolding of hen lysozyme. Biochemistry 37(23): 8473-8480.
-
Nishimura C, Dyson HJ, Wright PE (2002) The apomyoglobin folding pathway revisited: structural heterogeneity in the kinetic burst phase intermediate. J Mol Biol 322(3): 483-489.
-
Xie G, Timasheff SN (1997) The thermodynamic mechanism of protein stabilization by trehalose. Biophys Chem 64(1-3): 25-43.
-
Xie G, Timasheff SN (1997) Temperature dependence of the preferential interactions of ribonuclease A in aqueous co-solvent systems: thermodynamic analysis. Protein Sci 6(1): 222-232.
-
Xie G, Timasheff SN (1997) Mechanism of the stabilization of ribonuclease A by sorbitol: preferential hydration is greater for the denatured then for the native protein. Protein Sci 6(1): 211-221.
-
Privalov PL, Tiktopulo EI, Tischenko VM (1979) Stability and mobility of the collagen structure. J Mol Biol 127(2): 203-216.
- Sense, Gravity, Parity & Chirality in Mathematical Physics
- Quantum Lattice Simulations PHYSICS: Microcircuit Particle Formation and Observable Macroscopic Irreversible Time - A Discrete Lagrangian with Cellular Automata Framework
- Quantum Biology from Biomacromolecule to Cell, and Central Dogma Described by Quantum Theory
- Focus, Agility, Speed and Technology (FAST) for Sustainability and Growth
- Square Root Metric Geometry and Pati-Salam Model in Curved Space-Time
- A Simple System Demonstrating the Mpemba Effect in Classical Mechanics