Pretreatment with Nicotine Improves Behavioral Recovery and Ameliorates Neuroinflammation in a Rat Model of Spinal Cord Injury
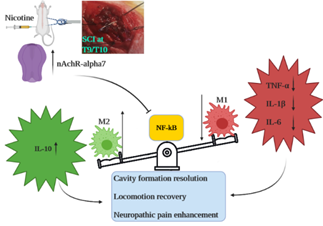
Background: This study addresses gaps in understanding the impact of nicotine on neuroinflammation, specifically in the context of secondary spinal cord injury (SCI). Uncertainties persist regarding the cellular and molecular components influenced by nicotine and whether its effects during the chronic phase of SCI are receptor-mediated. The research systematically investigates these aspects, aiming to provide insights into the nuanced mechanisms underlying nicotine’s impact on neuroinflammation in SCI. Methods: Male rats underwent either a sham operation or spinal cord injury (SCI). The study employed four nicotine groups and two methyllycaconitine (MLA) groups as treatment cohorts. Subsequent to behavioral assessments including locomotion impairment and mechanical allodynia, the optimal nicotine dosage was selected for comprehensive cellular and molecular evaluations of neuroinflammation in chronic phase of secondary SCI. Results: The administration of nicotine at a pre-treatment dosage of 1mg/kg demonstrated a mitigating effect on neuroinflammation, evidenced by improvements at both cellular and molecular levels. This intervention led to enhanced spinal cord tissue integrity, decreased M1/M2 polarization, and modulation of pro-inflammatory markers (NF-κB, TNF-α, IL-1β, IL-6), coupled with an increase in anti-inflammatory cytokine IL-10. Notably, these effects were negated upon the α7- nAChR, underscoring the receptor-dependent nature of nicotine’s impact. Conclusion: Pre-treatment with nicotine improved behavioral impairment of SCI possibly through alleviating neuroinflammation by changing macrophage phenotyping and cytokine levels. These effects were mediated by α7 nAChR.
Introduction
Spinal cord injury (SCI) is recognized as a multi- disciplinary and life-long condition. SCI has two different phases of injury. The primary SCI occurs at the moment of trauma as an inevitable event and it is defined as the mechanical injury of spinal cord. On the other hand, secondary SCI is characterized by series of molecular and cellular events which exacerbate the initial damage in a time dependent manner and also contributes to long-term behavioral impairment. Accordingly, neuroinflammation has been recognized as a crucial factor in the development of secondary SCI [1]. Neuroinflammation as the key component of secondary SCI encompasses time dependent interaction [2] between inflammatory cytokines and immune cells. Generally, inflammation per se is not inherently deleterious. In fact, inflammation plays a pivotal role in facilitating optimal healing process. To put it differently, inflammation within physiological parameters contributes to required healing process. Nonetheless, the pathological amplification and prolonged persistence of inflammation can compromise healing outcomes [3]. Evidentiary discourse underscores that the neuroinflammatory response precipitated by SCI predominantly manifests as an exacerbated inflammatory reaction during the acute post-injury phase, while a subsequent compromised healing responses and gliosis formation becomes conspicuous within the chronic phase ensuing injury [4]. Neuroinflammation with both molecular and cellular components assumes a crucial role in secondary SCI. While cellular elements incorporate the polarization of microglia/macrophage, molecular facets comprise notable markers such as NF-κB, TNF-α, IL-1β, and IL-6. It is firmly established that the classical polarization of microglia/ macrophage, denoted as the M1 pathway, culminates in the upregulation of CD86, NF-κB, TNF-α, IL-1β, and IL-6. Conversely, the alternative or anti-inflammatory polarization, designated as the M2 pathway, leads to the expression of CD163, CD206, and IL-10 [5].
Notably, interplay is evident between the molecular and cellular components of neuroinflammation. For instance, TNF-α not only intrinsically activates microglia via the classical pathway but also induces M1 macrophages to amplify TNF- α production. Moreover, it is important to underscore that the binary M1/M2 classification merely represents a simplified depiction of the dynamic cellular constituents in neuroinflammation. The actual scenario is far more intricate and exhibits temporal variability. In the aftermath of SCI induction, the balance between M1/M2 expression and their phenotypic profiles undergoes continuous transformation, aligning with the time-dependent nature inherent to neuroinflammation following SCI [6, 7, 8, 9]. Nicotine has demonstrated a significant impact on neuroinflammation and neuroprotection within diverse contexts, including cognitive impairment, ischemic stroke, neuropathic pain and also SCI. Prior research has demonstrated that the administration of nicotine is associated with a reduction in inflammatory markers such as NF-κB and TNF- α during the acute phase, as well as an amelioration of behavioral deficits in the chronic phase of secondary SCI [10, 11]. Nonetheless, two questions need to be addressed. First, it remains imperative to elucidate whether the effects of nicotine are receptor-mediated in the context of secondary SCI. Secondly, the relationship between behavioral improvements in the chronic phase and the neuroinflammatory status necessitates elucidation, especially given the paucity of results regarding the effects of nicotine on inflammation during the chronic phase of secondary SCI [12].
In the pursuit of elucidating these inquiries, it is worth noting that the neuroprotective and anti-inflammatory properties of nicotine within the central nervous system (CNS) have been posited to be orchestrated through the activation of the alpha-7 nicotinic acetylcholine receptor (α7 nAChR). This activation is postulated to yield a diminishment in the levels of inflammatory mediators such as NF-κB, TNF-α, IL-1β, and IL-6 at the molecular level, alongside the inhibition of classical macrophage/microglial polarization (M1 pathway) [13, 14]. A recent study has corroborated that the acute activation of α7 nAChR leads to an amelioration of behavioral deficits during the chronic phase of secondary Spinal Cord Injury (SCI), a phenomenon aligning with a shift in microglial polarization toward the M2 phenotype. As such, our hypotheses are anchored in the notion of receptor- mediated effects of nicotine, with a specific emphasis on the chronic evaluation of neuroinflammation [15]. In order to address the aforementioned questions, our investigative approach was twofold. Initially, we sought to ascertain the optimal dosage of nicotine with a particular focus on the assessment of behavioral outcomes. Subsequently, we assessed cellular aspects (i.e., macrophage polarization) and molecular components (specifically, NF-kB, TNF-α, IL-1β, and IL-6) of neuroinflammation on day 28 post-injury following acute nicotine treatment, while also investigating how these parameters might be influenced by selective blockade of the α7 nAChR.
Materials and Methods
Study Population
Sixty-four male Wistar rats, weighing 240–300 g, were provided from Tehran Pasteur Institute and were housed in separate cages that were equipped with free access to a sufficient amount of tap water and chow pellets. The room temperature was 23±2 °C with 50±5% humidity and a 12-hours light/dark cycle. The study protocol was executed in agreement with the National Institutes of Health (NIH) Guide for the Care and Use of laboratory animals (NIH publication No. 86-23, Eighth Ed.) and institutional and governmental concerns for animal care and use (Approval ID: IR.TUMS. MEDICINE.REC.1398.922). All possible efforts were made to minimize the study population and their suffering.
Drugs
Nicotine and a selective antagonist of α7 nAChR, Methyllycaconitine (MLA) were obtained from Sigma, St Louis, Missouri. Ketamine HCl (Gedeon Richter Ltd, Budapest, Hungary) and Xylazine HCl (Bayer, Leverkusen, Germany) were also purchased. Drugs were dissolved in physiologic saline and used through intraperitoneal injection.
Study Design
In a randomized setting, animals were assigned 8 to equal groups. Four groups received nicotine (Sigma-Aldrich, St. Louis, Missouri, United States) through intraperitoneal injection in the doses of 0.5, 1, 1.5, and 3 mg/kg in the attempt of identifying the amount with optimal behavioral outcomes, including locomotion and neuropathic pain recovery. 1.5 mg/ kg of MLA was administered intraperitoneally to one group while another group received the optimum dosage of nicotine in addition to 1.5 mg/kg of MLA. The timing of nicotine and MLA were 30 minutes and 45 minutes prior to the surgery, based on our pilot studies and previous publications [16]. The control and sham-operated groups merely received vehicle (saline 0.9%). All groups were followed for 28 days after surgery. In addition, rats that died with autophagia or urinary retention/infection were excluded. Four rats died of urinary retention/infection within ten days’ post SCI (one in control group, one in nicotine 3mg/kg treated group, one in MLA treated group and another in MLA + nicotine group). Moreover, two other rats were also excluded (one in control group and another in nicotine 1.5 mg/kg group) within a week following induction of SCI due to the autophagia and underwent sacrifice chamber. All data, including lab tests, locomotor, and sensory evaluations were collected by different observers, blinded to groups. All drugs were intraperitoneally administered 30 min prior to the surgery. Additionally, no sign of locomotor impairment caused by nicotine was observed in any group.
Surgical Procedure
A combination of 86 mg/kg Ketamine HCL and 13 mg/kg Xylazine HCL was injected intraperitoneally to anesthetize each animal. Dorsal hairs of the rodents were shaved with an electric razor, and the surgical site was cleaned with povidone-iodine. A prophylaxis injection of 30 mg/kg cefazolin was administered to all rodents before the beginning of the surgery. In the next step, each rat was located on a sterile, heating operating board in a prone position. After posterior midline incision and muscle dissection, complete laminectomy was operated on the T9/T10 vertebrae. The injury was induced on the spinal cord by compression with an aneurysmal clip (YASARGIL® Aneurysm clip system, Titanium mini clips FT712T; closing force, 110 g [1.08 N]; 4.7mm Blade length; 3.8mm maximum opening diameter) for 60 seconds. Finally, the wound sites were sutured, and animals were kept in a 35 °C incubator until recovery. The post-operative care was performed administrating normal saline (2 ml), cefazolin (20 mg/kg) and buprenorphine (0.1 mg/kg) for seven successive days. Voiding was performed twice a day manually until complete functional rehabilitation. Of note, the sham group underwent posterior midline incision without the induction of SCI.
BBB Assessment
The hindlimb locomotor function was assessed using the Basso, Beattie, Bresnahan (BBB) rating scale (Supplemental Table) in an open field at baseline and on days 1, 3, 7, 14, 21, and 28 after the surgery [17]. For each rat, the BBB score of hindlimbs were averaged and the result was documented. The assessor was blinded to the study groups. Score 21 indicates normal locomotion of hind limbs while 0 accounts for no locomotor function.
Von Frey Testing
For each rodent, the withdrawal thresholds of both left and right hind paws were assessed using the simplified up- down (SUDO) method of von Frey testing, which is valid and induces minimal stress to the rodents [18]. The assessments were conducted prior to SCI induction and on days 7, 14, 21, and 28 of the experiment. First, the rats were habituated to the test platform, which is an elevated box with a netting floor. Then, a series of von Frey hairs (a logarithmic series of 20 calibrated Semmes-Weinstein monofilaments; Stoelting Co., Wood Dale, IL) was smacked from below the platform to the central region of the plantar surface of the rat hind paws and were held for a maximum of 5 seconds. Prominent paw withdrawal or flinch was considered as positive responses. The start point of the test was with filament 10 (4.31, 2.0 g).
Sample Acquisition
At day 28 post-operation, half of the rats in each group were anesthetized with a mixture of 86 mg/kg Ketamine HCL and 13 mg/kg Xylazine HCL and then, 150 ml of phosphate buffered saline (PBS, pH 7.4) was perfused to their heart, followed by 250 ml of formalin-acid picric mixture (4% paraformaldehyde, 0.4% picric acid in 0.16 M phosphate buffer, pH 7.4) for fixation of the tissues. Three cm of the spinal cord was selected from the center of the injury site. Specimens were prepared and embedded in paraffin. The paraffinized specimens were sliced into samples with a thickness of 5 micrometers by a Leica 2135 microtome (Deerfield, IL, USA) for hematoxylin and eosin (H&E) and immunohistochemistry (IHC) assays. The other half of the rats underwent spinal cord dissection and the extracted samples were kept inside liquid nitrogen in a −70 °C freezer for Enzyme-Linked Immunosorbent Assay (ELISA) and qRT- PCR.
Histopathological Analysis
As mentioned above, half of the specimens were stained with H&E dye for histopathological assessment. The staining protocol is described in our previous publication [19]. After deparaffinization and rehydration of the specimens, the nuclei were stained by rinsing the slides in hematoxylin solution. In the next step, bluing was performed in 0.2% ammonia water. Then, the slides were counterstained in eosin-phloxine for 1 min and afterward samples were rinsed in 90%, 96% and 100% ethanol for 2 min. Finally, slides were embedded in xylene and mounting medium. The final products were examined using an optical microscope (resolution: 40×) to determine tissue and cavity percentages in the lesion sites. The Cavalieri’s method was used for calculation of sparred tissue and cavity percentages [20, 21].
IHC
IHC method was performed on half of the spinal cord specimens to evaluate the expression of the surface receptors of M1 and M2 macrophage/microglia, which are CD86 and CD206, respectively. The samples were rinsed by 0.1 M PBS in 4 steps. Afterward, in the attempt of retrieving the antigens, sections were placed in hydrochloric acid 2 normal (HCL) solutions for 30 min. Then, the effect of HCL was neutralized by borate buffer. In the next step, a 3% solution of Triton X-100 was used for 30 min to increase the cell membranes permeability. Goat serum (10%) was added to the background during a period of 30 min to block nonspecific reaction of the antibodies. Next, primary antibodies diluted with PBS (1:100) were added and the combination was transferred to a 2–8 °C refrigerator for 24 hours. Primary antibodies were as follows: mouse monoclonal anti-CD86 (ab220188, Abcam) for M1 and rabbit polyclonal anti-CD206 (ab64693, Abcam) for M2. Next, goat anti-rabbit antibody (Alexa Fluor® 488; ab150081, Abcam) and goat anti-mouse antibody (Alexa Fluor® 647; ab150115, Abcam) diluted with PBS (1:150) were added and the processed samples were incubated for 1.5 hours in a 37 °C incubator. Afterward, 4′,6-diamidino-2-phenylindole (DAPI) was added in a dim place to counterstain the nuclei. Notably, the samples were washed by PBS between the steps. Finally, the macrophages/ microglial were calculated, dividing prepared samples into 5 separate areas via an Olympus fluorescent microscope (×400) and images were taken from each area. The captured images were analyzed using the ImageJ software (Fiji version) [22]. The result was reported as the percentage of positive immunolabeled cells over the total cells in each selected region (the ratio number of positively stained cells/ total number of cells × 100).
qRT-PCR
Four main phases were conducted for qRT-PCR analysis of tumor necrosis factor-alpha (TNF-α), interleukin 6 (IL-6), interleukin 1 beta (IL-1β), interleukin 10 (IL-10), α7 nAChR, and NF-κB genes expression. First, total RNA was extracted from the specimens using Qiazol reagent (Qiagen, Germany). Second, one microgram of mRNA was reverse transcribed to cDNA according to manufacturer’s instructions (Fermentas, USA). Third, an Applied Biosystems 7300 Fast Real-Time PCR System with SYBR green PCR master mix (Applied Biosystems, CA, USA) was employed for qRT-PCR analysis. The primer sequences were designed (Primer3web version 4.1.0) and normalized with GAPDH as a control (Table 1). The thermocycler conditions were as follows: 95ºC for 15 min for activation of DNA polymerase, followed by 45 cycles of amplification at 94ºC for 15 sec, 60ºC for 15 secs and 72º for 30 sec. At last, melting curve analysis was conducted to confirm whether all primers provided a single PCR product. Sequences were normalized with GAPDH as a control. The 2−ΔΔCt method was used to measure the relative expression of aforementioned genes.
| Gene Name | Primers (Forward and Reverse) |
|---|---|
| α7 nAChR | F: AAAATGCCTAAGTGGACCAGAA |
| R: TCACTGCAGATCACCTCACTCT | |
| NF-κB | F: GTGCAGAAAGAAGACATTGAGGTG |
| R: AGGCTAGGGTCAGCGTATGG | |
| TNF-α | F: GCTCTTCTGTCTACTGAACTTCG |
| R: CAGCCTTGTCCCTTGAAGAGAA | |
| IL-1β | F: GATTGCTTCCAAGCCCTTGACT |
| R: AGGTGGAGAGCTTTCAGCTCA | |
| IL-6 | F: GGACTGATGTTGTTGACAGCC |
| R: GCTCTGAATGACTCTGGCTTTG | |
| IL-10 | F: AACTGCACCCACTTCCCAGT |
| R: ATGACAGCGTCGCAGCTGTAT | |
| GAPDH | F: TCAGAGCAAGAGAGGCATCC |
| R: GGTCATCTTCTCACGGTTGG |
Table 1: Primers for RT-PCR. Alpha-7 nicotinic acetylcholine receptor, α7 nAChR; Glyceraldehyde 3-phosphate dehydrogenase, GAPDH;
ELISA
Three pro-inflammatory factors, tumor necrosis factor- alpha (TNF-α), interleukin 1 beta (IL-1β), and interleukin 6 (IL-6), an anti-inflammatory mediator, interleukin 10 (IL- 10) and also protein levels of NF-κB and α7 nAChR were measured. The specimens were first homogenized in lysis buffer (2X Lysis Buffer; RayBiotech) and then centrifuged for 20 min at 13,000 rpm at 4 °C. Finally, the ELISA (Abcam, Cambridge, UK) method was employed to quantify the level of aforementioned proteins.
Statistical Analysis
All statistical analyses were performed using the SPSS software, version 25 (IBM Corp., Armonk, NY). The graphs were drawn using the GraphPad Prism software, version 7 (San Diego, CA, USA). The behavioral data were analyzed by the general linear model (GLM) repeated measures analysis followed by Tukey’s post hoc test. Besides, the one-way analysis of variance (ANOVA) test followed by Tukey’s post hoc test was employed to evaluate differences of histopathological scorings, pro-inflammatory and anti- inflammatory mediators, as well as α7 nAChR and NF-κB mRNA and protein expressions between the groups. The p values < 0.05*, < 0.01 and < 0.001* were accepted as statistically significant. Comparisons between two groups are reported as mean difference (95% confidence interval), which is abbreviated to MD (95% CI).
Results
Behavioral Assessment
Locomotion Impairment: According to locomotor assessment using the BBB scale, pre-operation scores were similar between the groups (p values > 0.05). One day after SCI induction, locomotor scores declined in all groups except for the sham-operated (Figure 1A). Repeated measure ANOVA analysis indicated significant effect of time [F (6, 336) = 4865, p < 0.001] and effect of the interaction between treatments and time [F (42, 336) = 120.2, p < 0.001] on locomotor scores during 28 days. Moreover, multiple comparisons using the Tukey as a post-hoc test exhibited the highest locomotor scores in rodents which received 1 mg/ kg of nicotine [nicotine (1 mg/kg) vs control: MD (95% CI) = 3.321 (2.45 to 4.19), p < 0.001]. Evidently, the BBB scores of all groups endured significantly lower comparing to sham (p values < 0.001). However, there was no significant difference between nicotine 1 mg/kg and nicotine 1.5 mg/kg treated groups (p = 0.736). Further comparisons between the groups are illustrated in Figure 1A & 1B.
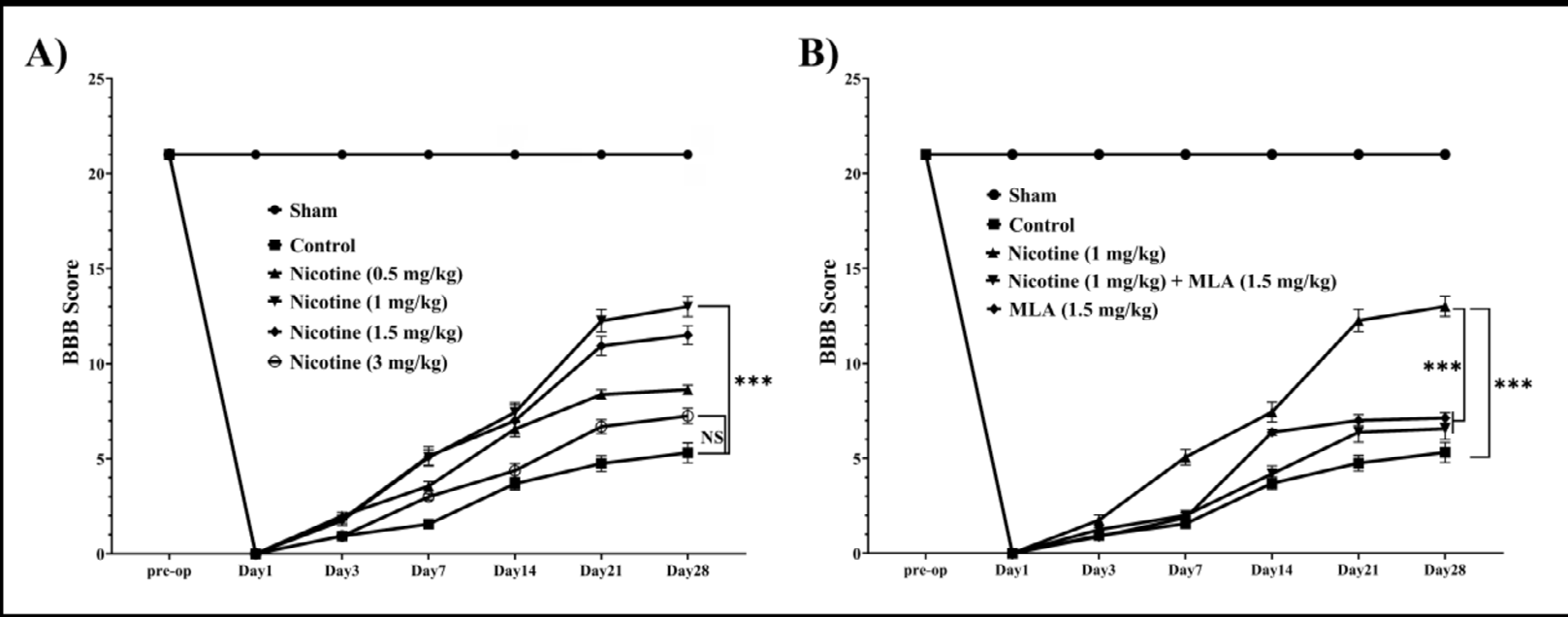
Figure 1: The presented figures depict the repeated measures of the Basso-Beattie-Bresnahan score, reflecting the locomotion status of animal subjects over a 28-day follow-up period (n=8). (A) Repeated measures delineate the efficacy of each nicotine treatment group, with nicotine 1 mg/kg exhibiting the most favorable impact on locomotion impairment. (B) Repeated measures further illustrate that the influence of nicotine on locomotion is contingent upon α7 nAChR mediation. Data are expressed as mean ± standard error mean (SEM). ***: p < 0.001.
Mechanical Allodynia: In order to assess mechanical allodynia, the von Frey method was performed weekly after induction of SCI and the withdrawal threshold was compared between the groups. Mechanical allodynia in sham-operated group remained intact while there was significant decline in the control group within one week after the surgery [MD (95% CI) = 54.25 (24.44 to 84.06), p < 0.001]. The decreasing trend of threshold to mechanical allodynia is evident from day 7 of the study (Figure 2A). Repeated measure analysis revealed significant effect of time [F (5, 280) = 1953, p < 0.001] and also effect of the time and treatment interaction [F (35, 280) = 38.31, p < 0.001], which represents different effectiveness of the administered treatments over time. Particularly, nicotine in doses of 1 mg/kg and 1.5 mg/kg could significantly rise the mechanical threshold compared to the control group on day 28 (p values = 0.009 and 0.025, respectively). Nevertheless, none of the groups achieved a mechanical resistance similar to the sham (Figure 2A).
![Figure 3: nicotine 1mg/kg vs nicotine 1mg/kg + MLA; MD (95% CI) = 6.28 (1.73 to 10.83), _p_ < 0.001].](/fulltextimages/11775/fig_3.png)
Figure 2: The figures above delineate repeated measures of Von Frey testing assessing mechanical allodynia status in animal subjects over a 28-day follow-up period (n=8). (A) The repeated measures illustrate the efficacy of each nicotine treatment group, with nicotine 1 mg/kg demonstrating an optimal effect on alleviating mechanical allodynia. (B) Further repeated measures indicate that the impact of nicotine on mechanical allodynia is contingent upon the mediation of the α7 nAChR. Data are presented as the mean ± standard error means (SEM). **: p < 0.01, *: p < 0.05.
Given that pre-treatment with a dosage of 1 mg/kg of nicotine demonstrated the most efficacious enhancement in mitigating behavioral deficits during the chronic phase of secondary SCI (Figures 1A, 2A), this specific dosage was chosen for subsequent investigations encompassing the analysis of cellular and molecular constituents involved in neuroinflammation on day 28. Remarkably, subsequent to the blockade of the α7 nAChR, a reversal of the observed trend in ameliorating behavioral deficits became evident, including evaluations related to locomotion (see Figure 1B, nicotine 1 mg/kg vs MLA + nicotine 1 mg/kg, p <0.001) and mechanical allodynia (see Figure 2B, nicotine 1 mg/kg vs MLA + nicotine 1 mg/kg, p = 0.05).
Cellular and Histology Assessment of Neuroinflammation
Spinal Cord Tissue Cavity Percentage: Figure 3 represents the mean cavity percentage at the site of injury 28 days after the induction of SCI. The one-way ANOVA test revealed significant difference between the groups [F (4, 25) = 114.8, p < 0.001]. According to the Tukey post-hoc test, SCI induction significantly the mean cavity percentage in the spinal cord tissue comparing to sham-operated group [MD (95%CI) = 28.52 (23.97 to 33.07), p value < 0.001]. However, treating spinal cord injured rats with 1 mg/kg of nicotine significantly reduced the cavity percentage compared to the control group [MD (95% CI) = 8.47 (3.92 to 13.02), p < 0.001]. Finally, blocking α7 nAChR with MLA led to reduction in promising effect of nicotine pre-treatment [see Figure 3: nicotine 1mg/kg vs nicotine 1mg/kg + MLA; MD (95% CI) = 6.28 (1.73 to 10.83), p < 0.001].
![Figure 4: The assessment of macrophages/microglia in the context of secondary SCI exhibits a multifaceted nature, characterized by a time- dependent progression. As anticipated, the initiation of SCI resulted in an augmented proclivity for macrophage polarization along the classic (M1) pathway [MD (95% CI) = 39.15 (32.56 to 45.73), _p_ < 0.001] and also decline toward the alternative (M2) pathway [MD (95% CI) = 32.71 (26.58 to 38.84), _p_ < 0.001] on day 28 following the injury. Pre-treatment with nicotine is associated with a notable reduction in M1 polarization [MD (95% CI) = 17.82 (11.23 to 24.41), _p_ < 0.001, Figure 4A] and a significant increase in M2 polarization [MD (95% CI) = 16.61 (10.48 to 22.74), _p_ < 0.001, Figure 4B]. Notably, the blocking of α7 nAChR by MLA exerts a reversing effect [see Figure 4B: nicotine vs nicotine + MLA, _p_ < 0.001], shifting the phenotypic balance towards a pro-inflammatory M1 polarization [Figure 4A: nicotine vs nicotine + MLA, _p_ < 0.01].](/fulltextimages/11775/fig_4.png)
Figure 3: Histopathological pictures of the groups, produced by H&E staining of spinal cord sections at the injury region after a period of 28 days: (A) sham, (B) Control, (C) Nicotine (1 mg/kg), (D) MLA (1.5 mg/kg), (E) Nicotine (1 mg/kg) + MLA (1.5 mg/kg). (F) Statistical analysis indicates the differences of cavity formation between the groups (mean ± standard deviation). Pre-treatment with nicotine improved cavity percentage at the site of injury. This effect was reversed following administration of MLA suggesting role of α7 nAChR : p < 0.01; *: p < 0.001.
Macrophage Polarization: The spinal cord phenotyping of M1 (CD86 in green) and M2 (CD206 in red) macrophages at the site of injury are depicted in Figure 4. The assessment of macrophages/microglia in the context of secondary SCI exhibits a multifaceted nature, characterized by a time- dependent progression. As anticipated, the initiation of SCI resulted in an augmented proclivity for macrophage polarization along the classic (M1) pathway [MD (95% CI) = 39.15 (32.56 to 45.73), p < 0.001] and also decline toward the alternative (M2) pathway [MD (95% CI) = 32.71 (26.58 to 38.84), p < 0.001] on day 28 following the injury. Pre-treatment with nicotine is associated with a notable reduction in M1 polarization [MD (95% CI) = 17.82 (11.23 to 24.41), p < 0.001, Figure 4A] and a significant increase in M2 polarization [MD (95% CI) = 16.61 (10.48 to 22.74), p < 0.001, Figure 4B]. Notably, the blocking of α7 nAChR by MLA exerts a reversing effect [see Figure 4B: nicotine vs nicotine + MLA, p < 0.001], shifting the phenotypic balance towards a pro-inflammatory M1 polarization [Figure 4A: nicotine vs nicotine + MLA, p < 0.01].
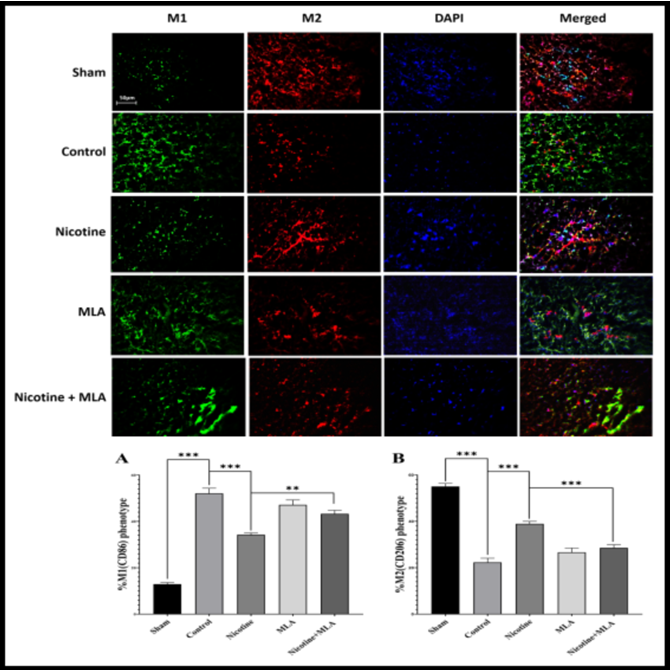
Figure 4: Representative immunofluorescent images featuring antibody staining depict M1 (CD86) and M2 (CD206) macrophage phenotyping at the injury site after a 28-day period (above). Diagrams below quantify the percentage of expression. A) SCI induced a shift towards M1 (CD86) polarization. Pre-treatment with nicotine mitigated M1 polarization, and this effect was found to be mediated by α7 nAChR. B) SCI resulted in a reduction of M2 polarization, which was ameliorated by nicotine pre- treatment. Administration of MLA indicated the involvement of α7 nAChR in this observed trend. Data are presented as the mean ± standard error means (SEM). *: p < 0.001, : p < 0.01, *: p < 0.05.
Molecular Assessment of Neuroinflammation: The NF- κB/TNF-α/IL-1β/IL-6 pro-inflammatory pathway plays a crucial role in persistent neuroinflammation following M1 macrophage polarization in secondary SCI. In contrast, IL- 10 is an important anti-inflammatory cytokine produced by M2 macrophages. Therefore, we aimed to evaluate the downstream molecular mediators of macrophage polarization in chronic phase of secondary SCI (Figure 5). Evidently, SCI significantly increased expression of NF-κB (p <0.001). Subsequent analysis revealed up-regulation of TNF-α/ IL-1β/IL-6 and down-regulation of IL-10 (p values < 0.001). Pre-treatment with nicotine resulted in mitigation of NF-κB/TNF-α/IL-1β/IL-6 inflammatory pathway (see Figure 5, NF-κB mRNA p <0.01, protein p <0.001; TNF-α p values <0.001; IL-1β p values <0.001; IL-6 p values <0.001) concomitant with an augmentation of IL-10 (Figure 5, mRNA p <0.001, protein p <0.01). Notably, the aforementioned impact of nicotine exhibited attenuation subsequent to the blockade of α7 nAChR (see Figure 5, NF-κB p values <0.01; TNF-α p values <0.001; IL-1β mRNA p <0.001, protein p <0.05; IL-6 mRNA p <0.01, protein p <0.001; IL-10 mRNA p <0.001, protein p <0.05). These findings imply that the influence of nicotine on molecular components of neuroinflammation was orchestrated by α7 nAChR.
![Figure 6: One-way ANOVA indicate significant difference between groups [mRNA: _F_ (3, 12) = 21.32, _p_ < 0.001; protein: _F_ (3, 12) = 115.5, _p_ < 0.001]. Secondary SCI led to significant down-regulation of α7 nAChR on day 28 post injury (_p_ values <0.001). Pre-treatment with nicotine 1mg/kg resulted in up-regulation of α7 nAChR [mRNA, Figure 6A: _p_ = 0.02; protein, Figure 6B: _p_ < 0.001]. Lastly, adding MLA to the pre- treatment reversed the effect of nicotine on the expression of this receptor significantly [mRNA, Figure 6A: _p_ = 0.027; protein, Figure 6B: _p_ < 0.01].](/fulltextimages/11775/fig_6.png)
Figure 5: The bar charts in this figure elucidate the mRNA expression (left column) and protein levels (right column) of NF-κB, TNF-α, IL-1β, IL-6, and IL-10 at the injury site after a 28-day period. Pre-treatment with nicotine elicited a down-regulation of pro-inflammatory markers (NF-κB, TNF-α, IL-1β, IL-6) and an up-regulation of the anti-inflammatory marker IL-10. Treatment with MLA revealed that these effects were mediated by the α7 nAChR. Data are presented as the mean ± standard error means (SEM). *: p < 0.001, : p < 0.01, *: p < 0.05.
Expression of α7 nAChR: The expression analysis of α7 nAChR regarding both mRNA and protein levels are depicted in Figure 6. One-way ANOVA indicate significant difference between groups [mRNA: F (3, 12) = 21.32, p < 0.001; protein: F (3, 12) = 115.5, p < 0.001]. Secondary SCI led to significant down-regulation of α7 nAChR on day 28 post injury (p values <0.001). Pre-treatment with nicotine 1mg/kg resulted in up-regulation of α7 nAChR [mRNA, Figure 6A: p = 0.02; protein, Figure 6B: p < 0.001]. Lastly, adding MLA to the pre- treatment reversed the effect of nicotine on the expression of this receptor significantly [mRNA, Figure 6A: p = 0.027; protein, Figure 6B: p < 0.01].
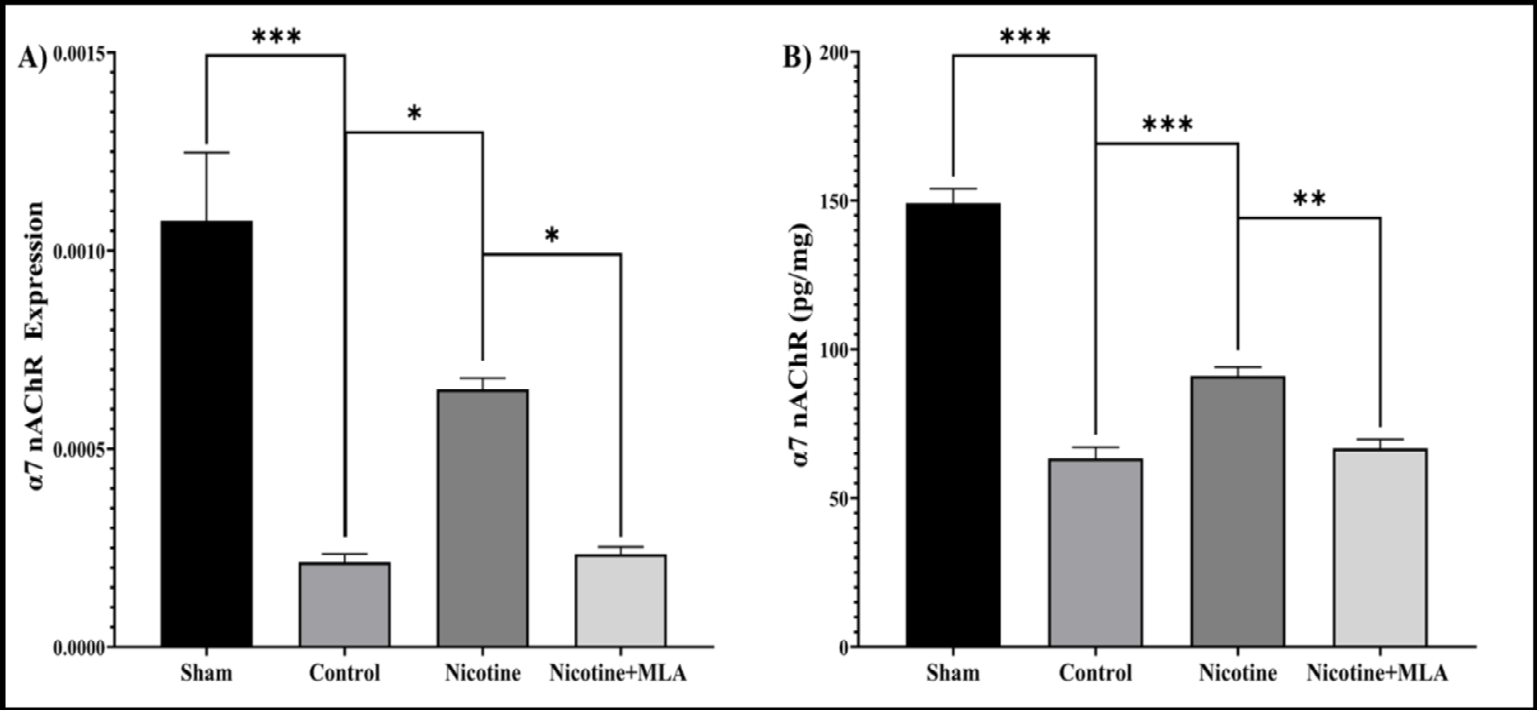
Figure 6: This figure portrays spinal cord mRNA (A) and protein (B) expression of the alpha7-nicotinic acetylcholine receptor (α7 nAChR) at the injury site over a 28-day period. Pre-treatment with nicotine yielded an up-regulation of the receptor, this effect subsequently reversed by treatment with methyllycaconitine (MLA). Data are presented as the mean ± standard error mean (SEM). *: p < 0.001, : p < 0.01, *: p < 0.05.
Discussion
Our behavioral assessments indicated that pre-treatment with nicotine improved secondary complications of SCI such as locomotor impairment and neuropathic pain. The nicotine alleviating effects appeared to be dose-dependent since 1mg/ kg of nicotine had the most notable impact on behavioral recovery, while 3mg/kg of the reagent apparently had less significant effect on locomotion and mechanical allodynia. Therefore, the optimal dosage of 1 mg/kg was chosen for molecular assay. Furthermore, nicotine demonstrated a mitigating effect on neuroinflammation at both the cellular and molecular levels. Nicotine induced a shift in M1/M2 macrophage polarization towards the alternative pathway and concurrently down-regulated the NF-κB/TNF-α/IL-1β/ IL-6 signaling cascade. Conclusively, the aforementioned effects were reverted subsequent to the administration of MLA, a selective antagonist of alpha7-nAchR. This outcome suggests that the mediation of these effects is attributable to the activity of the alpha7-nAchR receptor.
Neuro-inflammatory responses are integrated with secondary SCI, which is initiated in the first 24 hours after primary SCI and would last chronically. It is noteworthy that while acute inflammatory responses are considered as defensive mechanisms of the immune system against the damage, chronic neuroinflammation could compromise neuronal regeneration [23]. Neuroinflammation plays a critical role in the development of neuropathic pain and locomotor impairment, induced by SCI [24]. Although various mechanisms have been suggested to explain deficiencies after SCI, M1/M2 activity has been recently regarded as one of the main factors influencing neuroinflammation and axonal survival [25, 26, 27]{Walters, 2014 #232;Kong, 2017 #271}. In general, it seems that nervous-immune interactions take a remarkable part in the process of secondary SCI, which potentially could lead to significant positive and negative effects on neural survival and neural regeneration. In detail, M1/M2 macrophage/microglial polarization paradigm is acting as a seesaw in post-SCI neuroinflammation. The M1 phenotype, recognized as “classic macrophage/ microglial polarization”, produce inflammatory markers (e.g., TNF-α, IL-1β, IL-6) that bring about neurotoxic and inflammatory interactions, whereas the M2 phenotype, known as “alternative macrophage/microglial polarization”, tends to mediate neuro-regenerative responses through secretion of anti-inflammatory factors (e.g., IL-10). However, it appears that one-sided shift to M1 phenotype during the secondary SCI may facilitate undesirable outcomes varying from excessive inflammatory responses to fibrosis scarring [28]. Previous studies have confirmed this issue, showing that transplantation of M2 macrophages or manipulation of the existing microglial towards M2 phenotype could lessen neuroinflammation and histopathological abnormalities [6, 29, 30, 31, 32].
In this study, we aimed to elucidate the effects of α7 nAChR modification on neuroinflammation and histopathology in an animal model of SCI considering the role of M1/ M2 macrophage/microglial polarization paradigm. To the best of our knowledge, this is the first study that examines this relationship in SCI setting. α7 nAChR is a member of nicotinic acetylcholine receptors family and is expressed on both neuronal and non-neuronal cells, including endothelial cells, dendritic cells, macrophages, B cells and T cells. It has been well established that α7 nAChRs are involved in various inflammatory contexts such as sepsis, hemorrhage, rheumatoid arthritis, brain ischemia, myocardial infarction, Alzheimer’s disease, schizophrenia, pain and SCI [33, 34, 35, 36]. In addition, a remarkable number of studies have explored the effect of α7 in the extent of traumatic brain injury (TBI), which could be considered a close model to SCI. It has been shown that TBI could result in reduced levels of α7 nAChR in the injured region at both acute and chronic phases after the damage [37]. Besides, nicotine has been found effective in the improvement of neural loss and behavioral outcomes of subjects with TBI [38, 39]. The effects of α7 nAChR on regulation of inflammation in TBI models have been supported by observing that vagal nerve stimulation leads to the reduction of pro-inflammatory factors (TNF-α, IL-1β, IL- 6) through activation of the receptor [40, 41]. Various studies have revealed neuroprotective influences of α7 nAChR on the spinal cord. There is solid evidence concerning the stimulatory effects of α7 nAChR agonists on the level and activity of the receptor [34, 42, 43, 44, 45].
Rong, et al. [42] has indicated that pre-treatment with α2-adrenoreceptor selective agonist enhanced locomotor recovery after SCI among rats via amplification of α7 nAChR pathway [43]. Furthermore, it has been reported that α7 nAChR participates in determining the severity of neuropathic pain and mechanical allodynia. In this respect, Loram, et al. [46] demonstrated that both systemic and local administrations of α7 nAChR agonist could improve mechanical resistance to the forces, probably via reducing the spinal TNF-α level in a rat model of neuropathic pain [46, 47]. In two distinct studies, Ravikumar, et al. [10] showed that both single and multiple administrations of nicotine, a potent α7 nAChR agonist, attenuate oxidative stress, pro- inflammatory markers, and NF-κB activity while enhance sparring of spinal cord tissue and therefore, the recovery of SCI-induced locomotor dysfunction [10, 11].
Moreover, Richardson, et al. [48] indicated that nonsmokers with SCI experience lower grades of neuropathic pain after nicotine therapy [48]. Lee, et al. [3] suggested that nicotine may reduce inducible nitric oxide synthase (iNOS) protein and mRNA levels, probably due to the activation of α7 nAChR on microglial cells [12]. And finally, a recent study by Chen, et al. [15] has demonstrated that vagus nerve stimulation (VNS) ameliorated neuroinflammation through decreasing M1 and increasing M2 microglial polarization. This effect was in line with up-regulation of α7 nAChR while administration of MLA reversed this effect. Herein, we also detected significant improvement in functional and molecular status of the spinal cord injured rodents following administration of 1 mg/kg of nicotine, whereas 3mg/kg of nicotine appeared less beneficial. Interestingly, paradoxical effects of nicotine have been an issue in previous relevant studies and have been related to several features, including dosage, timing and selectivity [36, 49].
Beyond the single-dose therapy applied in the current study, investigating chronic nicotine injection could be an appropriate direction for future studies. There is mounting evidence that α7 nAChR by nicotine is associated with microglial activity. For instance, it was shown that pre- conditioning with nicotine prevented LPS-induced activated microglial cells from releasing pro-inflammatory cytokines and this effect was faded following the administration of MLA [50]. Several i_n vivo_ studies have also pointed to the anti-inflammatory effects of α7 nAChR on microglial cells. In a rat model of focal brain ischemia, Guan, et al. [51] discovered that nicotine regulates microglial proliferation and correspondingly the levels of TNF-α, IL-1β and neural loss even in the acute phase of post-injury and these effects were reversed by pre-treatment with α-bungarotoxin [51]. Several studies of CNS have exclusively discussed the role of α7 nAChR in the polarization status of M1/M2 macrophages/ microglia. In two recent studies of brain ischemia, it was noticed that activation of α7 nAChR is related to the inhibition of NF-κB, reduction of M1 markers (CD68, IL-1β, TNF-α, IL-6) and lesion volume, elevation of M2 markers (CD206, Arg-1, IL-10), as well as both short-term and long-term behavioral recoveries [52, 53]. Meanwhile, Ma, et al. [54] demonstrated that electro acupuncture therapy could enhance axonal survival via activation of α7 nAChR that leads to reduction of M1 markers and simultaneous elevation of M2 markers in a rat model of brain stroke [54].
There has been increasing interest regarding the role of M1/M2 polarization paradigm in mediation of secondary SCI, as well. A few experiments have targeted this theory to find novel therapeutic options for SCI. The shift of macrophages/ microglia to the M1 phenotype has been attributed to the activation of NF-κB in response to the binding of HMGB1 to TLRs. Results obtained from related investigations are consistent, suggesting higher levels of M1 markers (e.g., CD86) and lower levels of M2 markers (e.g., CD163, CD206) at the injury region of spinal cord. Observing higher concentrations of pro-inflammatory (e.g., TNF-α, IL-1β, IL- 6) and lower concentrations anti-inflammatory (e.g., IL-10) downstream markers confirms this alteration. Therefore, it is not surprising to detect neurodegenerative activities due to the established neuroinflammation [5, 28, 55]. Here, we consistently detected similar results in non-treated SCI rodents. Additionally, we discovered that elevation of α7 nAChR level due to nicotine therapy is correlated with shift reversion to M2 phenotype, probably through inhibition of NF-κB.
Conclusion
In summary, our findings propose that pre-treatment with nicotine ameliorates behavioral deficits in secondary SCI. This effect aligns with the concurrent improvement of cellular and molecular facets of neuroinflammation, including macrophage polarization and modulation of the NF-κB/TNF-α/IL-1β/IL-6 cascade. The pharmacological manipulation of α7 nAChR implies receptor-mediated effects of nicotine in this experimental context.
References
-
Anwar MA, Shehabi TSA, Eid AH (2016) Inflammogenesis of Secondary Spinal Cord Injury. Frontiers in cellular neuroscience 10: 98.
-
Nishimura S, Yasuda A, Iwai H, Takano M, Kobayashi Y, et al. (2013) Time-dependent changes in the microenvironment of injured spinal cord affects the therapeutic potential of neural stem cell transplantation for spinal cord injury. Molecular brain 6: 3.
-
Lee CYP, Chooi WH, Ng SY, Chew SY (2022) Modulating neuroinflammation through molecular, cellular and biomaterial‐based approaches to treat spinal cord injury. Bioengineering & Translational Medicine 8(2): e10389.
-
Hellenbrand DJ, Quinn CM, Piper ZJ, Morehouse CN, Fixel JA, et al. (2021) Inflammation after spinal cord injury: a review of the critical timeline of signaling cues and cellular infiltration. Journal of neuroinflammation 18(1): 1-16.
-
Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, et al. (2009) Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. The Journal of neuroscience 29(43): 13435-13444.
-
Shin T, Ahn M, Moon C, Kim S, Sim KB (2013) Alternatively activated macrophages in spinal cord injury and remission: another mechanism for repair? Molecular neurobiology 47(3): 1011-1019.
-
Pruss H, Kopp MA, Brommer B, Gatzemeier N, Laginha I, et al. (2011) Non-resolving aspects of acute inflammation after spinal cord injury (SCI): indices and resolution plateau. Brain pathology 21(6): 652-660.
-
Zhou X, He X, Ren Y (2014) Function of microglia and macrophages in secondary damage after spinal cord injury. Neural regeneration research 9(20): 1787-1795.
-
Gensel JC, Kopper TJ, Zhang B, Orr MB, Bailey WM (2017) Predictive screening of M1 and M2 macrophages reveals the immunomodulatory effectiveness of post spinal cord injury azithromycin treatment. Scientific reports 7: 40144.
-
Ravikumar R, Flora G, Geddes JW, Hennig B, Toborek M (2004) Nicotine attenuates oxidative stress, activation of redox-regulated transcription factors and induction of proinflammatory genes in compressive spinal cord trauma. Brain research Molecular brain research 124(2): 188-198.
-
Ravikumar R, Fugaccia I, Scheff SW, Geddes JW, Srinivasan C, et al. (2005) Nicotine attenuates morphological deficits in a contusion model of spinal cord injury. Journal of neurotrauma 22(2): 240-251.
-
Lee MY, Chen L, Toborek M (2009) Nicotine attenuates iNOS expression and contributes to neuroprotection in a compressive model of spinal cord injury. Journal of Neuroscience research 87(4): 937-947.
-
Tyagi E, Agrawal R, Nath C, Shukla R (2010) Inhibitory role of cholinergic system mediated via alpha7 nicotinic acetylcholine receptor in LPS-induced neuro- inflammation. Innate immunity 16(1): 3-13.
-
Cui WY, Li MD (2010) Nicotinic modulation of innate immune pathways via alpha7 nicotinic acetylcholine receptor. Journal of neuroimmune pharmacology: The official journal of the Society on NeuroImmune Pharmacology 5(4): 479-488.
-
Chen H, Feng Z, Min L, Deng W, Tan M, et al. (2022) Vagus nerve stimulation reduces neuroinflammation through microglia polarization regulation to improve functional recovery after spinal cord injury. Frontiers in Neuroscience 16: 813472.
-
Kasbi AA, Haddadi NS, Dehdashtian A, Afshari K, Jazaeri SZ, et al. (2019) Acute Activation of α7-Nicotinic Receptors by Nicotine Improves Rodent Skin Flap Survival Through Nitrergic System. Annals of Plastic Surgery 83(2): 211-216.
-
Basso DM, Beattie MS, Bresnahan JC (1995) A sensitive and reliable locomotor rating scale for open field testing in rats. Journal of neurotrauma 12(1): 1-21.
-
Bonin RP, Bories C, Koninck YD (2014) A simplified up-down method (SUDO) for measuring mechanical nociception in rodents using von Frey filaments. Randomized Controlled Trial 10: 26.
-
Moradi K, Golbakhsh M, Haghighi F, Afshari K, Nikbakhsh R, et al. (2020) Inhibition of phosphodiesterase IV enzyme improves locomotor and sensory complications of spinal cord injury via altering microglial activity: Introduction of Roflumilast as an alternative therapy. Int Immunopharmacol 86: 106743.
-
Abdanipour A, Tiraihi T, Taheri T, Kazemi H (2013) Microglial activation in rat experimental spinal cord injury model. Iran Biomed J 17(4): 214-220.
-
Michel RP, Cruz-Orive LM (1988) Application of the Cavalieri principle and vertical sections method to lung: Estimation of volume and pleural surface area. J Microsc 150(2): 117-136.
-
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, et al. (2012) Fiji: an open-source platform for biological-image analysis. Nature methods 9(7): 676- 682.
-
Beck KD, Nguyen HX, Galvan MD, Salazar DL, Woodruff TM, et al. (2010) Quantitative analysis of cellular inflammation after traumatic spinal cord injury: evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain: A journal of neurology 133(2): 433-447.
-
Rice T, Larsen J, Rivest S, Yong VW (2007) Characterization of the early neuroinflammation after spinal cord injury in mice. Journal of neuropathology and experimental neurology 66(3): 184-195.
-
Walters ET (2014) Neuroinflammatory contributions to pain after SCI: Roles for central glial mechanisms and nociceptor-mediated host defense. Experimental neurology 258: 48-61.
-
Kong X, Gao J (2017) Macrophage polarization: A key event in the secondary phase of acute spinal cord injury. Journal of cellular and molecular medicine 21(5): 941- 954.
-
Li J, Liu Y, Xu H, Fu Q (2016) Nanoparticle-Delivered IRF5 siRNA Facilitates M1 to M2 Transition, Reduces Demyelination and Neurofilament Loss, and Promotes Functional Recovery After Spinal Cord Injury in Mice. Inflammation 39(5): 1704-1717.
-
David S, Kroner A (2011) Repertoire of microglial and macrophage responses after spinal cord injury. Nature reviews Neuroscience 12(7): 388-399.
-
Guerrero AR, Uchida K, Nakajima H, Watanabe S, Nakamura M, et al. (2012) Blockade of interleukin-6 signaling inhibits the classic pathway and promotes an alternative pathway of macrophage activation after spinal cord injury in mice. Journal of neuroinflammation 9(1): 40.
-
Ma SF, Chen YJ, Zhang JX, Shen L, Wang R, et al. (2015) Adoptive transfer of M2 macrophages promotes locomotor recovery in adult rats after spinal cord injury. Brain, behavior, and immunity 45: 157-170.
-
Nakajima H, Uchida K, Guerrero AR, Watanabe S, Sugita D, et al. (2012) Transplantation of mesenchymal stem cells promotes an alternative pathway of macrophage activation and functional recovery after spinal cord injury. Journal of neurotrauma 29(8): 1614-1625.
-
Ren Y, Young W (2013) Managing inflammation after spinal cord injury through manipulation of macrophage function. Neural plasticity 2013: 945034.
-
Cai B, Chen F, Ji Y, Kiss L, Jonge WJ, et al. (2009) Alpha7 cholinergic-agonist prevents systemic inflammation and improves survival during resuscitation. Journal of cellular and molecular medicine 13(9b): 3774-3785.
-
Bertrand D, Lee CH, Flood D, Marger F, Donnelly-Roberts D (2015) Therapeutic Potential of alpha7 Nicotinic Acetylcholine Receptors. Pharmacological reviews 67(4): 1025-1073.
-
Bencherif M, Narla ST, Stachowiak MS (2014) Alpha7 neuronal nicotinic receptor: A pluripotent target for diseases of the central nervous system. CNS & neurological disorders drug targets 13(5): 836-845.
-
Bencherif M, Lippiello PM, Lucas R, Marrero MB (2011) Alpha7 nicotinic receptors as novel therapeutic targets for inflammation-based diseases. Cellular and molecular life sciences 68(6): 931-949.
-
Kelso ML, Oestreich JH (2012) Traumatic brain injury: central and peripheral role of alpha7 nicotinic acetylcholine receptors. Current drug targets 13(5): 631-636.
-
Verbois SL, Hopkins DM, Scheff SW, Pauly JR (2003) Chronic intermittent nicotine administration attenuates traumatic brain injury-induced cognitive dysfunction. Neuroscience 119(4): 1199-1208.
-
Guseva MV, Hopkins DM, Scheff SW, Pauly JR (2008) Dietary choline supplementation improves behavioral, histological, and neurochemical outcomes in a rat model of traumatic brain injury. Journal of Neurotrauma 25(8): 975-983.
-
Kox M, Pompe JC, Pickkers P, Hoedemaekers CW, Vugt ABV, et al. (2008) Increased vagal tone accounts for the observed immune paralysis in patients with traumatic brain injury. Neurology 70(6): 480-485.
-
Wang H, Yu M, Ochani M, Amella CA, Tanovic M, et al. (2003) Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421(6921): 384-388.
-
Akaike A, Takatori YT, Kume T, Izumi Y (2010) Mechanisms of neuroprotective effects of nicotine and acetylcholinesterase inhibitors: Role of alpha4 and alpha7 receptors in neuroprotection. Journal of molecular neuroscience 40(1-2): 211-216.
-
Rong H, Zhao Z, Feng J, Lei Y, Wu H, et al. (2017) The effects of dexmedetomidine pretreatment on the pro- and anti-inflammation systems after spinal cord injury in rats. Brain, behavior, and immunity 64: 195-207.
-
Costantini A, Viola N, Berretta A, Galeazzi R, Matacchione G, et al. (2018) Age-related M1/M2 phenotype changes in circulating monocytes from healthy/unhealthy individuals. Aging 10(6): 1268-1280.
-
Jonnala RR, Buccafusco JJ (2001) Relationship between the increased cell surface α7 nicotinic receptor expression and neuroprotection induced by several nicotinic receptor agonists. Journal of Neuroscience research 66(4): 565-572.
-
Loram LC, Harrison JA, Chao L, Taylor FR, Reddy A, et al. (2010) Intrathecal injection of an alpha seven nicotinic acetylcholine receptor agonist attenuates gp120-induced mechanical allodynia and spinal pro- inflammatory cytokine profiles in rats. Brain, behavior, and immunity 24(6): 959-967.
-
Loram LC, Taylor FR, Strand KA, Maier SF, Speake JD, et al. (2012) Systemic administration of an alpha-7 nicotinic acetylcholine agonist reverses neuropathic pain in male Sprague Dawley rats. The Journal of Pain 13(12): 1162- 1171.
-
Richardson EJ, Ness TJ, Redden DT, Stewart CC, Richards JS (2012) Effects of nicotine on spinal cord injury pain vary among subtypes of pain and smoking status: results from a randomized, controlled experiment. The journal of pain: Official journal of the American Pain Society 13(12): 1206-1214.
-
Oloris SC, Abel AAF, Jubala CM, Fosmire SP, Helm KM, et al. (2010) Nicotine-mediated signals modulate cell death and survival of T lymphocytes. Toxicology and applied pharmacology 242(3): 299-309.
-
Shytle RD, Mori T, Townsend K, Vendrame M, Sun N, et al. (2004) Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. Journal of neurochemistry 89(2): 337-343.
-
Guan YZ, Jin XD, Guan LX, Yan HC, Wang P, et al. (2015) Nicotine inhibits microglial proliferation and is neuroprotective in global ischemia rats. Molecular neurobiology 51(3): 1480-1488.
-
Han Z, Shen F, He Y, Degos V, Camus M, et al. (2014) Activation of α-7 nicotinic acetylcholine receptor reduces ischemic stroke injury through reduction of pro- inflammatory macrophages and oxidative stress. PloS one 9(8): e105711.
-
Han Z, Li L, Wang L, Degos V, Maze M, et al. (2014) Alpha-7 nicotinic acetylcholine receptor agonist treatment reduces neuroinflammation, oxidative stress, and brain injury in mice with ischemic stroke and bone fracture. Journal of neurochemistry 131(4): 498-508.
-
Ma Z, Zhang Z, Bai F, Jiang T, Yan C, et al. (2019) Electroacupuncture Pretreatment Alleviates Cerebral Ischemic Injury Through α7 Nicotinic Acetylcholine Receptor-Mediated Phenotypic Conversion of Microglia. Frontiers in cellular neuroscience 13: 537.
-
Gensel JC, Zhang B (2015) Macrophage activation and its role in repair and pathology after spinal cord injury. Brain research 1619: 1-11.
- Gallic and Citric Acid Present in the Peels of Tropical Fruits as an Alternative in the Fight against Cancer
- Treating the Forehead Lines with Combination of Forehead and Glabellar Botulinum Toxin Among Japanese Patients
- Clinical Evaluation of Patients Suffering from Breast Cancer & Determination of Treatment Therapies and Better Strategies Related to Breast Cancer
- Medieval Recipes by Al-Zahrāwī for Heart Palpitations Treatment
- Etiology and Prescription Errors of Myocardial Infarction in Different Health Care Systems of Azad Kashmir
- Early Diagnosis and Multidisciplinary Management of Turner Syndrome: A Paediatric Case Study