An Unusual Case of Sarcoma Developing in a Previously Diagnosed Ependymoma-Case Report
Ependymomas are rare glial tumors arising from the ependymal cells lining the ventricles of central nervous system. It accounts for only 1.8% of all adult brain tumors. Gliosarcoma is a rare primary brain tumour composed of both malignant glial and sarcomatous components. Under the WHO classification, conventional gliosarcomas are considered a subtype of glioblastoma, an astrocytic tumor. We present a case of sarcoma developing in a patient previously diagnosed as ependymoma. Both the initial supratentotrial ependymoma and subsequent sarcoma are thought to have type 1 C11orf95-RELA fusion. This conclusion is supported by the finding of identical genetic alterations in both tumor elements. The term ependymosarcoma was first coined by Rodriguez F, et al. who described 11 cases of ependymal tumors with sarcomatous features. To date, 19 cases of ependymal tumors classified as WHO Grade II or III with sarcomatous changes have been published in the literature, with 10 of these cases occurring at recurrence. Genetic and mutational analyses have shown that identical genetic mutations occur in both the glial and sarcomatous tissues. This indicates that, though histologically distinct, the two components have a common genetic origin, favoring a monoclonal origin for both components
Introduction
Ependymomas are rare glial tumors arising from the ependymal cells lining the ventricles of central nervous An Unusual Case of Sarcoma Developing in a Previously Diagnosed Ependymoma-Case Report system. It accounts for only 1.8% of all adult brain tumors [1]. They may develop supratentorially, infratentorially and in spinal cord. These tumors arising from different locations are genetically distinct and represent distinct tumour entities. Ependymal tumors are classified into four histopathological subtypes including subependymoma (grade I), myxopapillary ependymoma (grade I), ependymoma (grade II), ependymoma, RELA Clin Radiol Imaging J
fusion-positive (grade II or III), and anaplastic ependymoma (grade III) according to the World Health Organization (WHO) Classification of Tumours of the Central Nervous System [2]. Various forms of differentiation occur in ependymomas, such as lipomatous, cartilaginous and osseous, melanotic, eosinophilic and, most recently described, neuronal [3].
Gliosarcoma is a rare primary brain tumor composed of both malignant glial and sarcomatous components. Under the WHO classification; conventional gliosarcomas are considered a subtype of glioblastoma, an astrocytic tumor. We present a case of sarcoma developing in a patient previously diagnosed as ependymoma. Both the initial supratentotrial ependymoma and subsequent sarcoma is thought to have type 1 C11orf95–RELA fusion [4]. This conclusion is supported by the finding of identical genetic alterations in both tumor elements [3]. However, sarcomatous change in oligodendroglial tumors has been previously reported and there have been isolated case reports of the same phenomenon occurring in ependymomas and subependymomas [3]. This unique finding strongly suggests that sarcoma likely developed from the malignant transformation of the primary ependymoma.
Case Report
A fifty one year old female presented with occipital headache and diminished vision in both eyes. MRI of brain had showed right occipital space occupying lesion. She underwent gross total excision of the lesion in Jan, 2014 and was diagnosed as grade II right occipital ependymoma, showing positivity for Glial Fibrillary Acidic Protein (GFAP), Epithelial Membrane Antigen (EMA) and negative for cytokeratin and MIB-1 on immunohistochemistry (Figures 1 & 2).
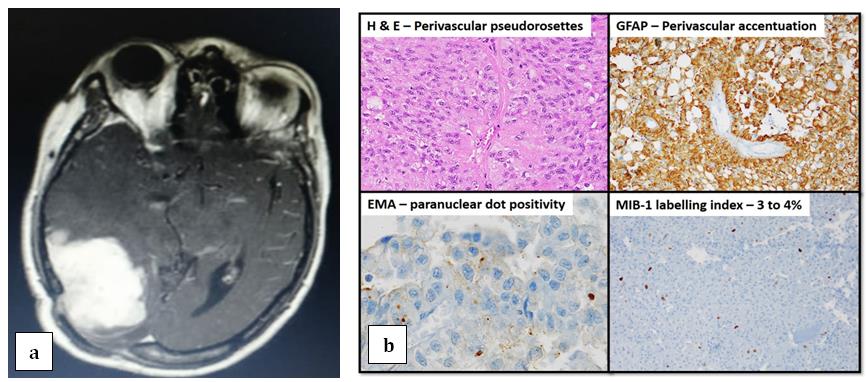
She was disease free during regular follow up with clinical examination and imaging. Four years later, she developed similar complaints headache and blurring of vision predominantly in the left eye. MRI of brain showed fairly well defined lobulated extra axial T2W/FLAIR hyperintense, T1W hypointense lesion showing intense homogenous enhancement and adjacent dural tail, measuring 39x57x52mm in right parietal region suggestive of recurrence. She underwent right parieto- occipital craniotomy and excision of the lesion in August, 2018. The post-operative histopathology report now revealed round to polyhedral cells having moderate granular eosinophilic cytoplasm, pleomorphic hyperchromatic nucleus and prominent nucleoli suggestive of undifferentiated sarcoma-high grade in nature (Figures 2a-2c).
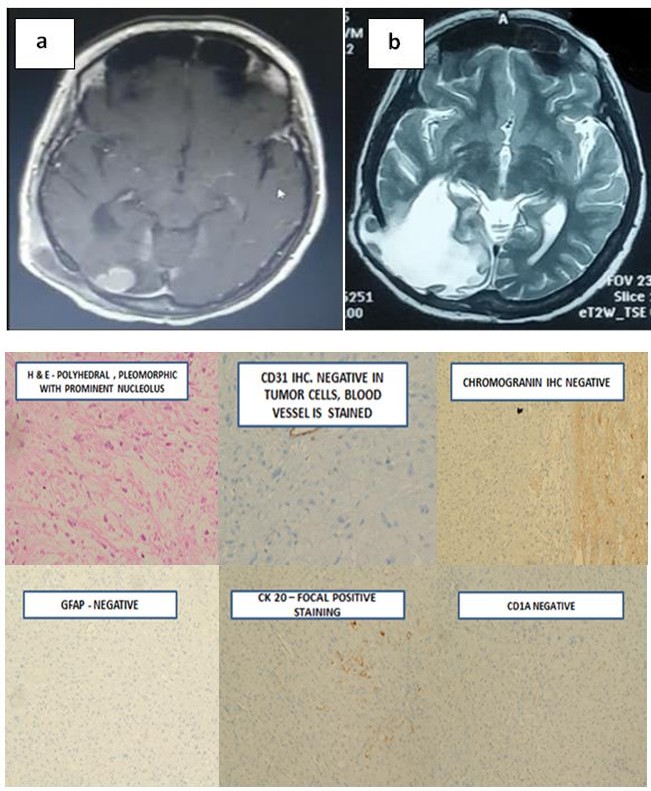
Figures 2A & B: a) Post gadolinium enhanced T1 weighted image showing recurrence in the occipito-parietal region surrounded by edema b) T2 weighted post gadolinium contrast weighted image showing edema in the post operative site.
Figure 2c: Microscopically revealed round to polyhedral cells having moderate granular eosinophilic cytoplasm, pleomorphic hyperchromatic nucleus and prominent nucleoli, GFAP negative, CD 31 negative CD1a negative, CK 20 focal positive staining on immunohistochemistry, suggestive of undifferentiated sarcoma-high grade in nature.
dose of radiation therapy and tolerated the treatment fairly well. She is symptom free and stable response at 6months follow up and kept under close observation.
Discussion
Ependymomas are a group of glial tumors that usually arise within or adjacent to the ependymal lining of the ventricular system and are thought to be derived from the radial glial cells in the subventricular zone [5]. Ependymomas most commonly occur in the posterior fossa, in contact with the fourth ventricle, or in the intramedullary spinal cord; also they occasionally occur in the brain parenchyma outside the posterior fossa, and very rarely outside the central nervous system (CNS). The cellularity and architecture of ependymoma vary considerably, not only from case to case, but also from area to area within the same tumor. Furthermore, the cells comprising ependymomas vary from glial to epithelial in their appearance. Ependymal tumors are graded into four histopathological subtypes including subependymoma (grade I), myxopapillary ependymoma (grade I), ependymoma (grade II), ependymoma, RELA fusion-positive (grade II or III), and anaplastic ependymoma (grade III) according to the World Health Organization (WHO) Classification of Tumours of the Central Nervous System [2].
So far, the glial fibrillary acidic protein and vimentin are often used for distinguishing ependymoma from other tumors of the CNS. Epithelial membrane antigen (EMA) is another marker that has been proven to be highly selective in ependymoma and is considered a marker for the differentiated forms of these tumors [6]. Due to their glial nature, ependymomas usually display GFAP immunoreactivity in pseudorosette pattern. Recent molecular analyses strongly suggest that although ependymomas from the supratentorial compartment, infratentorial region, and spinal cord have similar histologic features, biologically they are distinct, possibly explaining the regional differences in prognosis. Molecular and genetic data demonstrate distinct expression profiles and DNA copy number alterations. Importantly, Parker M, et al. recently described a novel oncogenic fusion on chromosome 11 involving a previously uncharacterized gene, C11orf95 and RELA, a transcriptional factor in the NF-kB pathway. This fusion transcript was estimated to occur in two-thirds of supratentorial ependymomas [7].
Gliosarcoma is a neoplasm with glial and mesnchymal components. It is considered to be a subtype of glioblastoma. It accounts for 2% of glioblastomas. Sarcomatous component of gliosarcoma shows a wide range of morphologic variations such as chondroid, rhabdomyoblastic, osseous, lipomatous and leiomyomatous. Although majority of the gliosarcomas the glial component isastrocytic, on a rare occasion it can be oligodendroglial. Sarcomatous metaplasia in ependymomal is rare phenomenon. The term ependymosarcoma was first coined by Rodriguez F, et al. who described 11 cases of ependymal tumors with sarcomatous features [3]. To date, 19 cases of ependymal tumors classified as WHO Grade II or III with sarcomatous changes have been published in the literature, with 10 of these cases occurring at recurrence. The histogenesis of ependymosarcomas, especially the sarcomatous component, is controversial [8]. This controversy extends also to gliosarcomas where initially the mesenchymal component was thought to have arisen from malignant transformation of the proliferative vasculature of astrocytomas. However, genetic and mutational analyses have shown that identical genetic mutations occur in both the glial and sarcomatous tissues. This indicates that, though histologically distinct, the two components have a common genetic origin, favoring a monoclonal origin for both components [4].
The relationship between the glial and sarcomatous elements in gliosarcomas has been subject to speculation for many years now. Although initially thought to be secondary to neoplastic transformation of the often proliferative vasculature of high-grade astrocytomas, molecular genetic studies have demonstrated identical aberrations in the glial and sarcomatous components. Applied methods have included interphase cytogenetics, comparative genomic hybridization, and mutation analysis of p16, PTEN, and TP53. All studies to date have concluded that the glial and sarcomatous tissues are genetically related in most instances. In our case FISH could not be performed due to financial constraints. Amplification of MDM2 has been described in ependymomas and conventional gliosarcomas, in some instances in the sarcomatous component only or with co amplification of CDK4 [3]. Because sarcomatous transformation may be found in astrocytic as well as oligodendroglial and ependymal tumors, it suggests that this phenomenon might be analogous to sarcomatoid change in a variety of carcinomas occurring outside of the central nervous system [9, 10, 11].
Conclusion
Genetic and mutational analyses have shown that identical genetic mutations occur in both the glial and sarcomatous tissues. This indicates that, though histologically distinct, the two components have a common genetic origin, favoring a monoclonal origin for both components [4]. In conclusions, in this report, we explore the rare occurrence of sarcomatous change in ependymal tumor. Although the present WHO classification assigns gliosarcomas to the astrocytic category, diagnosticians must be aware that ependymal and oligodendroglial tumors also may be site of sarcomatous change [3].
References
-
CBTRUS (2011) Statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2004-2007.
-
Fukuoka K, Kanemura Y, Shofuda T, Fukushima S, Yamashita S, et al. (2018) Significance of molecular classification of ependymomas: C11orf95-RELA fusion-negative supratentorial ependymomas are a heterogeneous group of tumors. Acta Neuropathologica Commun 6(1): 134.
-
Rodriguez F, Scheithauer B, Perry A, Oliveira A, Jenkins R, et al. (2008) Ependymal Tumors With Sarcomatous Change (Ependymosarcoma): A Clinicopathologic and Molecular Cytogenetic Study. Ame J Surgical Pathol 32(5): 699-709.
-
Cachia D, Wani K, Penas-Prado M, Olar A, McCutcheon I, et al. (2014) C11orf95–RELA fusion present in a primary supratentorial ependymoma and recurrent sarcoma. Brain Tumor Pathology 32(2): 105-111.
-
Shuangshoti S, Rushing E, Mena H, Olsen C, Sandberg GD (2005) Supratentorial extraventricular ependymal neoplasms: a clinicopathologic study of 32 patients. Cancer 103(12): 2598-2605.
-
Hasselblatt M, Paulus W (2003) Sensitivity and specificity of epithelial membrane antigen staining patterns in ependymomas. Acta Neuropathol 106(4): 385-388.
-
Parker M, Mohankumar KM, Punchihewa C, Weinlich R, Dalton JD, et al. (2014) C11orf95–RELA fusions drive oncogenic NF-κB signalling in ependymoma. Nature 506(7489): 451-455.
-
Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, et al. (2015) CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008-2012. Neuro Oncology 17(4): 1- 62.
-
Alotaibi FE, Petrecca K (2013) Short Interval Infield Sarcoma Development following Resection of Glioblastoma and Adjuvant Radiotherapy and Temozolomide. Case Reports Medicine 2013: 4.
-
Stenzel AE, Fenstermaker RA, Wiltsie LM, Moysich KB (2019) Disparities among racial/ethnic groups of patients diagnosed with ependymoma: analyses from the Surveillance, Epidemiology and End Results (SEER) registry. J Neurooncol 144(1): 43-51.
-
Pakos EE, Goussia AC, Pitouli EJ, Tsekeris PG (2004) Primary cerebral gliosarcoma: a case presentation with review of the literature. J BUON 9(4): 481-484.
- Ultrasound Guided Therapeutic Nerve Blocks
- Cyclops Lesion Without ACL Reconstruction: A Rare Case in a Patient with Intact Anterior Cruciate Ligament and Tibial Plateau Fracture
- Dosimetric Comparison between Two Dose Calculation Algorithms in SBRT Treatment of Lung Cancer in Ring-based and C-arm Radiation Therapy Equipment
- Adolescent Testicular Adrenal Rest Tumors: A Case Report and Review of the Literature
- Giant Intrathoracic Lipoma: A Rare Presentation
- Image of a Right Renal Angiomyolipoma Complicated by Hemorrhage