Endocrine System Related to Diabetes Mellitus
Diabetes mellitus has a complicated and poorly understood pathogenesis. However, it turns out to be an abnormal metabolic state linked to systemic vascular bed injury. Additionally, the endocrine system is not functioning normally in people with diabetes mellitus, according to the body of data. Uncertainty surrounds the relationship between the alterations in the endocrine system and the hormonal environment caused by aberrant glucose and fat metabolism, decreased insulin action, or both. A review of the literature shows that the entire endocrine system, including the hormones produced by the hypothalamus, pituitary, adrenal, thyroid, and parathyroid glands as well as the vitamin D system, the gonads, and the adipose tissue, is dysfunctional. Some of these abnormalities may be corrected with proper metabolic management and insulin therapy. It is still unknown how much these endocrine system changes contribute to the vascular pathologies seen in people with diabetes mellitus and whether some of the abnormalities seen in the endocrine system are due to a fundamental cellular defect in the diabetic syndrome. A complex, long-term metabolic condition called diabetes mellitus causes increased blood glucose levels as a result of insufficient insulin synthesis or uptake. The goal of this review article is to give a thorough overview of the complex interactions between the endocrine system and diabetes mellitus pathogenesis. It is still unknown how much these endocrine system changes contribute to the vascular pathologies seen in people with diabetes mellitus and whether some of the abnormalities seen in the endocrine system is due to a fundamental cellular defect in the diabetic syndrome. A complex, long-term metabolic condition called diabetes mellitus causes increased blood glucose levels as a result of insufficient insulin synthesis or uptake. The goal of this review article is to give a thorough overview of the complex interactions between the endocrine system and diabetes mellitus pathogenesis. It results from dysregulation of the endocrine system, primarily involving the pancreas, and its hormones, particularly insulin and glucagon. This review article provides an in-depth analysis of recent advances in our understanding of the endocrine system’s; role in diabetes mellitus, encompassing both type 1 and type 2 diabetes. The review begins by elucidating the pivotal roles of the endocrine system in maintaining glucose homeostasis, highlighting the pancreas; contribution through the secretion of insulin and glucagon. It explores the etiological factors that underlie diabetes, including genetics, lifestyle, and environmental influences. The article then delves into the pathophysiological mechanisms responsible for the onset and progression of diabetes mellitus, encompassing insulin resistance, β-cell dysfunction, and the involvement of various hormones, cytokines, and adipocytes. A large worldwide health burden is posed by diabetes mellitus, a set of metabolic illnesses characterized by increased blood glucose levels. The endocrine system, which is made up of numerous glands and hormones, is intricately related to the aetiology Diabetes & Obesity International Journal 2 Bhavana U and Punna V. Endocrine System Related to Diabetes Mellitus. Diabetes Obes Int J 2023, 8(4): 000279. Copyright© Bhavana U and Punna V. of diabetes and plays a crucial role in regulating glucose metabolism. The goal of this in-depth review article is to give a complete analysis of the state of knowledge at the moment about how the endocrine system and diabetes mellitus interact. Beginning with a discussion of the physiological part played by the endocrine system in maintaining glucose homeostasis, the overview highlights the important hormones insulin, glucagon, and others as well as the major participants in this process. After that, it explores the complex aetiology of diabetes mellitus, including type 1, type 2, and gestational diabetes, highlighting the role that genetic, environmental, and lifestyle variables have in the progression of the disease. Advancements in the understanding of the endocrine system’s; intricate signalling pathways, including insulin secretion, insulin resistance, and hormonal regulation, are explored in detail.
Introduction
Endocrine System
The term “endocrine” implies that in response to specific stimuli, the products of these glands are hormones. A plethora of hormones regulate over all body’s functions. Communication among various regions of the body is essential for organism to respond appropriately to any changes in the internal and external environments i.e. ensured by neuroendocrine system. The endocrine system plays a critical role in regulating various physiological processes in the human body, including blood glucose levels. Endocrine system consists of a network of glands that produce and release hormones into the bloodstream. These hormones act as messengers, regulating a wide range of bodily functions, such as metabolism, growth, immune response, and energy balance. In the context of diabetes, two key hormones are of particular importance: insulin and glucagon [1].
Insulin: Produced by the beta cells of the pancreas, insulin is a hormone that plays a central role in regulating blood sugar levels. When you eat, your blood sugar (glucose) levels rise, and insulin is released to help cells absorb glucose for energy or storage. Insulin ensures that blood sugar levels remain within a narrow, healthy range. Glucagon: Produced by the alpha cells of the pancreas, glucagon has the opposite effect of insulin. When blood sugar levels are low, for example, between meals or during physical activity, glucagon is released to signal the liver to release stored glucose into the bloodstream, raising blood sugar levels.
Diabetes Mellitus
A heterogeneous, complicated metabolic condition called diabetes is characterized by increased blood glucose levels brought on by either resistance to the action of insulin, inadequate insulin secretion, or both. Type 1 diabetes, Type 2 diabetes, and gestational diabetes are the three most widely used classifications. The symptoms of type 2 diabetes (T2DM) include insulin resistance and a relative lack of insulin production [2].
Even if the absolute plasma insulin concentration (during fasting and after meals) is typically higher, it is typically insufficient to sustain healthy glucose homeostasis” relative”: to the degree of insulin resistance. In the majority of T2DM patients, insulin secretion capability gradually declines over time. Diabetes mellitus is a group of physiological dysfunctions characterized by hyper-glycaemia resulting directly from insulin resistance, inadequate insulin secretion, or excessive glucagon secretion [3].
Type 1 Diabetes (T1D): Typically diagnosed in childhood or young adulthood, T1D results from an autoimmune reaction that destroys the insulin-producing beta cells in the pancreas. People with T1D require lifelong insulin therapy to survive.
- Is destruction of pancreatic beta cells.
- It is auto immune disorder Type 2 diabetes (T2D): The most common form, T2D is often linked to insulin resistance and lifestyle factors. It can develop at any age but is more common in adults. It may be managed through lifestyle changes, medication, or insulin.
• It is progressively impaired glucose regulation due to a combination of dysfunctional pancreatic beta cells and insulin resistance (Figure 1).
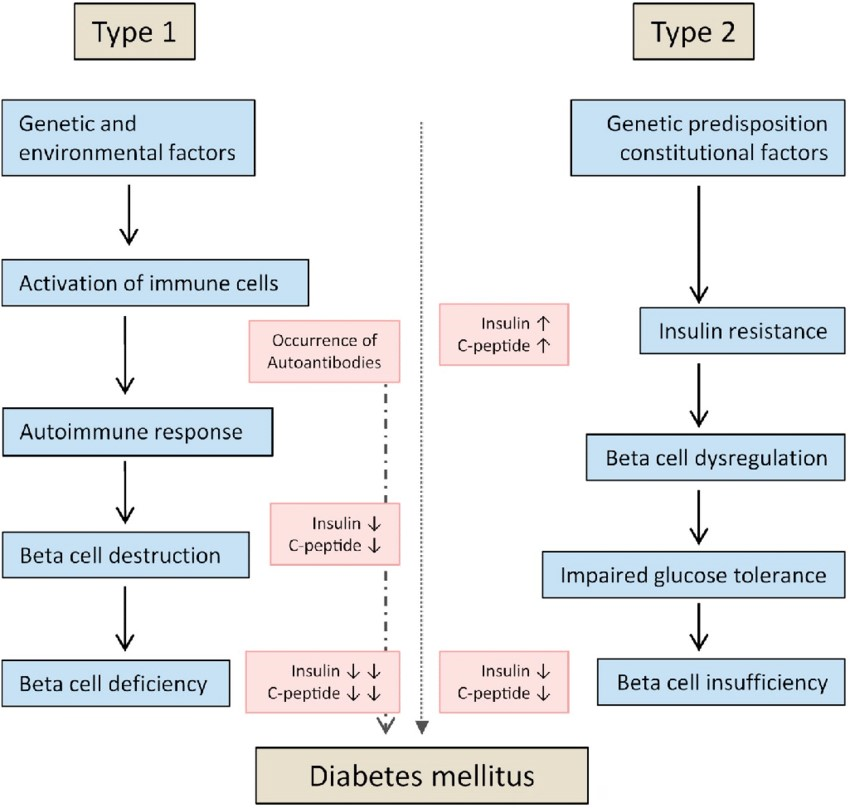
Mainly associated impairment in:
Pancreas
Insulin: Helps control carbohydrate metabolism (i.e., lowers blood sugar levels). Insulin plays a critical role in maintaining stable blood sugar levels, preventing hyperglycaemia (high blood sugar) and hypoglycaemia (low blood sugar) [4]. Glucagon: Helps control carbohydrate metabolism (i.e., increases blood sugar levels). Glucagon, plays a vital role in maintaining stable blood sugar levels, ensuring that the body has a constant supply of glucose for energy (Figure 2).
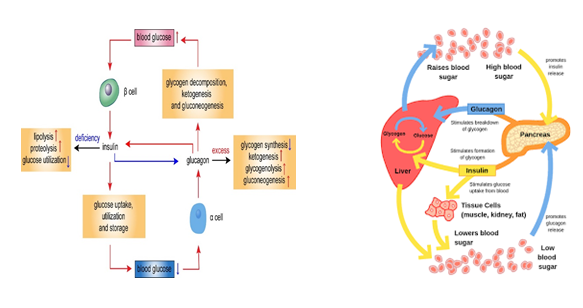
Endocrine System, Hormones and Glands Associated to Diabetes Mellitus
Pituitary Gland
- Location: Pituitary gland is located at the base of your brain, behind the bridge of your nose and directly below your hypothalamus. It sits in an indent in the sphenoid bone called the sella turcica [5].
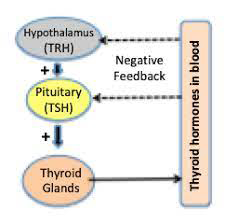
- Role: The pituitary gland, or hypophysis, is the ‘master gland that secretes multiple hormones which regulate the functioning of other endocrine organs, such as the thyroid, adrenal cortex and gonads (Figure 3).
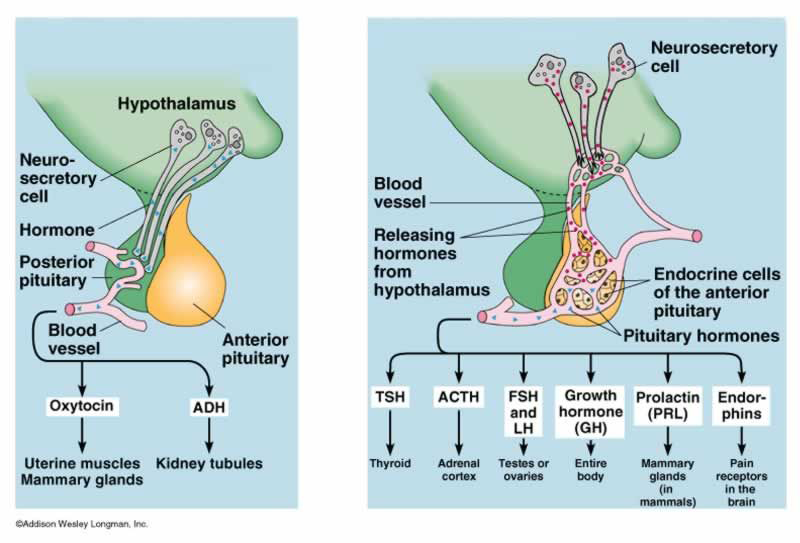
Role in Diabetes Mellitus
The anterior and posterior hypophyseal hormones alter glucose metabolism in health and disease. Secondary diabetes may occur due to hypersecretion of anterior pituitary hormones like adrenocorticotrophic hormone in Cushing’s disease and growth hormone in acromegaly. Other hormones like prolactin, gonadotropins, oxytocin and vasopressin, though not overtly associated with causation of diabetes, have important physiological role in maintaining glucose homeostasis. Hypoglycemia is not an unusual occurrence in hypopituitarism. Many of the medications that are used for treatment of hypophyseal diseases alter glucose metabolism. Agents like pasireotide should be used with caution in the setting of diabetes, whereas pegvisomant should be given preference [6].
Diabetes mellitus itself, on the other hand, can alter the functioning of hypothalamic pituitary axis; this is documented in both type 1 and type 2 diabetes.
Though none of the major pituitary hormones directly the endocrine glandular components [7].
Glucose Metabolism in Anterior Pituitary Disease
Hyperfunction: This encompasses four recognized conditions of hormone hypersecretion:
- Pituitary Adenoma Secreting Growth Hormone (GH), or Acromegaly: Pituitary adenoma secreting: GH enhances hepatic glucose production by opposing the action of insulin on the liver and stimulates gluconeogenesis. GH also increases hepatic and peripheral insulin resistance. One of the mechanisms behind peripheral insulin resistance is substrate competition with glucose due to higher free fatty acid availability from GH induced lipolysis, On the other hand IGF-1 improves peripheral insulin sensitivity but is not enough to override the diabetogenic action of chronic GH excess. Studies have demonstrated the association of IGF-1 levels Ana DM as predictors of mortality in acromegaly [8].
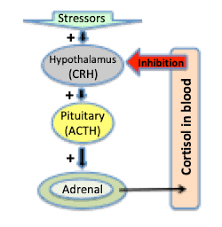
- Adrenocorticotrophic Hormone (ACTH), or Cushing’s Disease: Cushing’s disease, or ACTH secreting pituitary adenoma, is another recognized cause of DM. The prevalence of DM in Cushing’s disease is 40-45% and an additional 10-30% have impaired glucose tolerance. A state of chronic cortisol excess stimulates hepatic gluconeogenesis partly resulting from increased mobilization of gluconeogenic substrate from peripheral tissues [9]. Cortisol also has a permissive effect on glucagon and epinephrine in inducing gluconeogenesis and glycogenolysis. Insulin secretory defects coupled with increased peripheral insulin resistance have additional role in development of hyperglycemia (Figure 4).
Prolactin
Milk synthesis depends on prolactin in a crucial way. Serum prolactin levels and milk production are both decreased in postpartum rats when diabetes is induced. Additionally, diabetic rats had lower prolactin response to sucking. Interestingly, milk production is fully restored in diabetic rats despite the fact that insulin therapy does not normalize serum prolactin levels [10]. In non-pregnant diabetic mice, circulating prolactin levels remain unchanged, but prolactin responses to various stimuli, such as stress and TRH, are reduced. Prolactin levels in type 1 and type 2 diabetic men are normal or even raised, according to human research, but they are decreased in type 1 diabetic women. Poorly controlled type 1 diabetes patients have aberrant and decreased circadian patterns of plasma prolactin, which may indicate hypothalamic dysfunction (172, 173, or diminished pituitary responses to TRH). Diabetes patients’ aberrant prolactin production may be caused by reduced insulin- mediated prolactin gene expression [11]. The mean serum prolactin levels in people with type 1 and type 2 diabetes are the same, and there is no association between serum prolactin levels and the duration of diabetes or levels of glycosylated hemoglobin. Together, it is uncertain if diabetes mellitus has an impact on prolactin function that is clinically relevant [12, 13, 14, 15]. Impotence is not expected to result from changes in prolactin levels in diabetes mellitus. Additionally, neither type of diabetic man’s prolactin levels nor their potential for sex are correlated. Prolactin-secreting pituitary adenoma; and very rarely Prolactin Hypersecretion. Unlike acromegaly and Cushing’s disease, hyperprolactinemia is not a recognized cause of DM. On one hand, prolactin has been shown to induce beta-cell growth in the pancreas [16, 17], while on the other hand, prolactin is also hypothesized to increase insulin resistance by blocking adiponectin and interleukin (L)-6 production in fat cells.
Thyroid-Stimulating Hormone (TSH)
It secreting pituitary adenoma and Gonadotrophin- secreting adenomas are usually clinically non-functioning but secretion of intact follicle-stimulating hormone (FSH) in women of reproductive age group has been associated with ovarian hyperstimulation. Thyrotoxicosis can cause insulin resistance and predispose to diabetes. TSH secreting pituitary adenoma by producing thyrotoxicosis can worsen glucose intolerance like in any other thyrotoxicosis state, but the condition is very rare and as per our knowledge, diabetes arising from this condition has not been reported. Gonadotropin-secreting tumors are non-functional and in exceptional circumstances can produce manifestations related to gonadotropin excess and not usually linked to glucose homeostasis [18] (Figure 5).
Hypofunction: Pituitary hypofunction or hypopituitarism implies the deficiency of one or more anterior and posterior pituitary hormones. Hypoglycemia can be a presenting feature of hypopituitarism. It results primarily from hypocortisolism, secondary to defect in ACTH secretion coupled with deficiency of GH [19].
GH deficiency has a different impact on glucose homeostasis at various stages of life. Children with GH deficiency have a tendency to be insulin sensitive and can develop spontaneous fasting hypoglycemia due to reduced hepatic glucose production. GH deficient adults who are not receiving GH substitution, however, are insulin resistant. Besides overt hormonal deficiency, functional hypogonadism presumed to be due to a non-organic defect in hypothalamic- pituitary-testicular axis, is a common cause of low plasma testosterone in adult males with comorbidities like diabetes and obesity. Low plasma total testosterone levels have been linked with insulin resistance, impaired glucose tolerance, higher visceral adipose tissue and adverse cardiovascular (CV) outcomes. Whether metabolic syndrome is the cause behind hypogonadism or its effect remains an area of controversy (Figure 6).
Glucose Metabolism in Posterior Pituitary Disease
Oxytocin: An imbalance in Oxytocin secretion related to a disease process is not known to cause any specific defect in glucose metabolism; however, over last few years the physiological role of oxytocin in glucose and lipid metabolism has been recognized. Oxytocin preferentially suppresses intake of sweet-tasting carbohydrates [20]. Oxytocin levels were found to be lower in newly diagnosed, T2DM and obese individuals. There is some research suggesting a potential link between, Oxytocin and diabetes mellitus, but it’s important to note that the relationship is complex and not fully understood. Oxytocin may influence glucose metabolism and insulin sensitivity. Some studies have explored the use of oxytocin in the treatment of diabetes to improve insulin sensitivity (Figure 7).
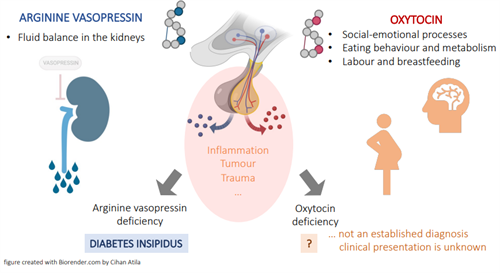
Vasopressin: In reaction to hyperosmolarity, hypovolemia, and stress, vasopressin is produced in the hypothalamus and released into the blood by the neurohypophysis. Plasma vasopressin levels are increased in both poorly controlled type 1 diabetic individuals and animals with diabetes, most likely as a result of the hypovolemia brought on by hyperglycemia. Although diabetic rats have a higher basal discharge rate of vasopressinergic neurons, the response to osmotic stimulation is very moderate. The lack of circadian vasopressin secretion observed in rats with uncontrolled diabetes mellitus may be explained by this slowed vasopressin response, which may indicate an osmoregulation malfunctionn. Depending on how long a person has been in a hyperglycemic condition, their ability to respond to While fast insulin withdrawal in type 1 diabetes causes an unchanged or an increased osmoreceptor sensitivity during the hyperglycemic state, it is reported that the sensitivity is reduced in newly diagnosed untreated hyperglycemic individuals. The integrity of the autonomic nervous system, sodium levels, volume status, the effects of nonosmotic stimuli, and other variables all need to be considered when interpreting these findings.
Patients with uncontrolled type 2 diabetes have decreased or depleted levels of vasopressin storage in the posterior lobe of the pituitary. Through glycemic control improvements, this is partially reversed. O smotic stimuli with vasopressin is affected. There are two types of vasopressin receptors: V1 and V2. Vasopressin’s antidiuretic impact is mediated by V2 receptors, which are found in the kidney’s collecting duct. Vasopressin’s effects on the vascular tissue and hepatic glycogenolysis are mediated by V1 receptors, which are found in the kidney, brain, and blood vessels. Vasopressin levels are higher and renal V1 receptors are downregulated in hyperglycemic conditions. Both the antidiuretic V2 receptor and its second-messenger system activity do not exhibit this (Figure 8).
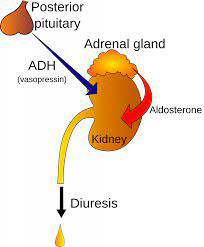
Vasopressin may be a factor in the complications of diabetes. It might be involved in diabetic nephropathy’s early stages. It has been shown that type 2 diabetic patients had lower urine levels of aquaporin 2, the vasopressin-sensitive water channel that promotes tubular water reabsorption, than controls. With better diabetes control, this impairment can be corrected. As a result, the acquired form of relative nephrogenic diabetes insipidus may contribute in part to the polyuria observed in the hyperglycemic state, which is characterized by a reduction in the capacity to concentrate urine.
However, the primary cause of the polyuria observed in hyperglycemic conditions is still glycosuria secondary to increased glucose load [21].
Growth Factor Axis with Insulin-Like Growth Hormone
Growth hormone (GH) inhibits insulin action, increases lipolysis, and plays a vital role in protein synthesis. IGF-1, a strong growth and differentiation factor, is also a major mediator of many of its actions. The expression of the genes for pituitary GH and GH receptor, hypothalamus somatostatin and GH-releasing hormone (GHRH), and GH is downregulated in animal diabetes. Growth hormone levels in the blood are suppressed, and the amplitude, length, and frequency of GH pulses are all reduced. In humans, type 1 diabetes that is not well controlled is linked to high plasma levels of GH and low levels of IGF-1. Additionally, strict metabolic regulation does not worsen the elevated mean 24-hour GH levels. Additionally, type 1 diabetics have higher urine GH excretion, and the urinary GH concentration is correlated with the daily dose of insulin. It’s interesting to note that in people with type 1 diabetes, glucose fails to lower GH levels and may even, paradoxically, increase them. Additionally, even in the absence of concurrent hypoglycemia, GH levels can increase during insulin treatment. Uncontrolled type 1 diabetics have been demonstrated to have a blunted IGF-1 response and an increased GH response to exogenous GHRH, indicating a central hyper-sensitivity to GHRH and a peripheral resistance to GH. Hepatic growth hormone receptors (GHR) downregulation may contribute to the GH resistance state. Additionally, as type 1 diabetics have insufficient portal insulinization and hepatic IGF-1 production and secretion, insulin is required for both processes (Figure 9).
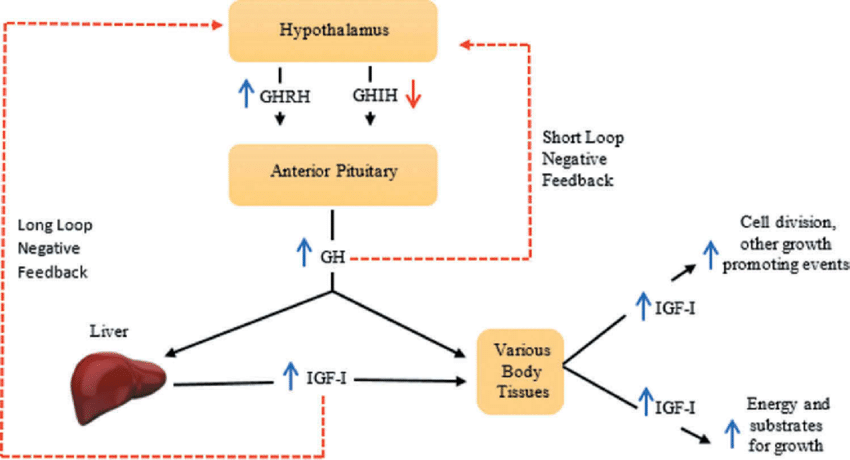
In type 1 diabetes, the IGF-1 level is lowered, and insulin treatment partially corrects this abnormality. Additionally, type 1 diabetes reduces IGF-1 activity, and the level of activity is correlated with the degree of glycemic control. IGF-1 levels in type 1 diabetics may rise as a result of better glycemic management, according to some research, but not others. IGF-1 levels are also reduced in type 2 diabetics. Low levels of GH-binding protein (GHBP) and liver GHR gene expression are caused by low insulin levels. The injection of insulin corrects these abnormalities. Patients with type 1 diabetes and type 2 diabetes who have low insulin levels also have lower GHBP levels. The degree of insulinogenic is correlated with the levels of GHBP.
Glucocorticoids in the Hypothalamic-Pituitary- Adrenocortical Axis
In both animal and human diabetes mellitus, the secretion of corticotrophin-releasing hormone (CRH) is inversely correlated. In STZ diabetic rats, the expression of the hypothalamic CRH gene is diminished; this expression is restored by insulin therapy. Other hypothalamic variables, in addition to CRH, may activate the hypothalamic-pituitary- adrenal axis in uncontrolled diabetes and indirectly decrease the expression of the CRH gene through a negative glucocorticoid feedback. Basal serum CRH levels are lower and basal serum adrenocorticotropic hormone (ACTH) levels are higher in type 2 diabetic individuals. Atypical cortisol and ADH circadian rhythms are also linked to type 2 diabetes. It is not CRH stimulation that is responsible for this rise in plasma ACTH and cortisol in type 2 diabetes patients. In this sense, insulin may promote both peripheral and central hypothalamic-pituitary-adrenocortical secretory activity in humans. Hypercortisolism and diabetes mellitus are connected. Patients with type 2 diabetes have raised 24-hour plasma cortisol profiles, higher nocturnal plasma cortisol rises, and higher fasting serum cortisol levels. Cortisol production may be increased by poor glycemic management. Patients with type 2 diabetes also have lessened dexamethasone-induced suppression of cortisol metabolites. These alterations in cortisol metabolism could be a factor in the hypertension, obesity, and menstrual irregularities that are frequent in type 2 diabetes and are also present in Cushing’s syndrome. The reaction of ACTH and cortisol to hypoglycemia is reduced by strict glycemic management. Rather than an alteration in the way the adrenal glands operate, this may be the result of a hypothalamic-pituitary response to hypoglycemia. The treatment of diabetic individual’s may benefit from this phenomenon.
The Adrenal Glands and Their Hormones
Location: The adrenal glands are small structures located on top of the kidneys. Structurally, they consist of an outer layer (i.e., the cortex) and an inner layer (i.e., the medulla). The adrenal cortex produces numerous hormones, primarily corticosteroids (i.e., glucocorticoids and mineralocorticoids). The cortex is also the source of small amounts of sex hormones; those amounts, however, are insignificant compared with the amounts normally produced by the ovaries and testes [22].
The adrenal medulla generates two substances— adrenaline and noradrenaline—that are released as part of the fight-or-flight response to various stress factors. The primary glucocorticoid in humans is cortisol (also called hydro-cortisone), which helps control carbohydrate, protein, and lipid metabolism. For example, cortisol increases glucose levels in the blood by stimulating gluconeogenesis in the liver and promotes the formation of glycogen (i.e., a molecule that serves as the storage form of glucose) in the liver. Cortisol also reduces glucose uptake into muscle and adipose tissue, thereby opposing the effects of insulin. Furthermore, in various tissues, cortisol promotes protein and lipid breakdown into products (i.e., amino acids and glycerol, respectively) that can be used for gluconeogenesis. Adrenal Medulla: The body’s reaction to hypoglycemia is significantly influenced by the release of epinephrine from the adrenal medullary. A quick increase in adrenal medullary secretion occurs when the glucose concentration falls below overnight fasting levels because of regulating glucose-sensitive neurons in the central nervous system. Increasing hepatic glucose output and providing a substitute substrate in the form of free fatty acids, this increase—which may reach levels 25–50 times above baseline—suppresses insulin release and prevents insulin-mediated glucose uptake in muscles. The function of the adrenal medulla may be impacted by diabetes.
The weight and size of the adrenal medulla rise along with the concentration of the adrenal hormones adrenaline, norepinephrine, and dopamine in experimental diabetes. Patients with type 1 diabetes have higher 24-hour urine epinephrine excretion, which is only partially reversed by better glycemic management. The diurnal regularity of epinephrine secretion is maintained despite an increase in the mean daily amount. Autopsies of patients with type 1 diabetes revealed fibrosis and medullitis of the adrenal medulla, and there was a correlation between the degree of fibrosis and the length of the diabetes. Type 2 diabetes did not exhibit this medullary fibrosis. Even before the development of the clinical diabetes, circulating anti-adrenal-medullary antibodies were seen in new-onset type 1 diabetic individuals. Autonomic neuropathy and long-term diabetic patients may not show the normal rise in circulating epinephrine levels in response to hypoglycemia. This impairment is significant in the pathophysiology of the hypoglycemia unawareness state seen in people with long-term type 1 diabetes, together with a faulty glucagon response. Strict glycemic control is linked to a decrease in the plasma glucose threshold that initiates adrenaline release. Animals with diabetes have significantly reduced adrenal gland epinephrine release in response to direct electrical stimulation of the splanchnic nerve terminals. Physiologic alterations in reaction to catecholamine’s are also linked to experimental diabetes. Adrenergic receptors in the myocardium decreased, and catecholamine’s had a stronger vasoconstrictive effect on blood vessels. It has been suggested that the higher prevalence of hypertension associated with diabetes may be partially explained by the enhanced catecholamine vasoconstrictive action.
Diabetes either increases the lipolytic response to epinephrine or has no effect on it.
Kidneys
Renin–Angiotensin–Aldosterone System: The renin- angiotensin-aldosterone system (RAAS) is crucial for maintaining electrolyte balance and cardiovascular health. Renin is released by renal juxtaglomerular cells, which sets off a chain of events that leads to the production of angiotensin II. By increasing glomerular capillary permeability, stimulating aldosterone secretion, and stimulating the synthesis of the presclerotic cytokine transforming growth factor (TGF- ), angiotensin II causes systemic and intraglomerular hypertension (Figure 10).
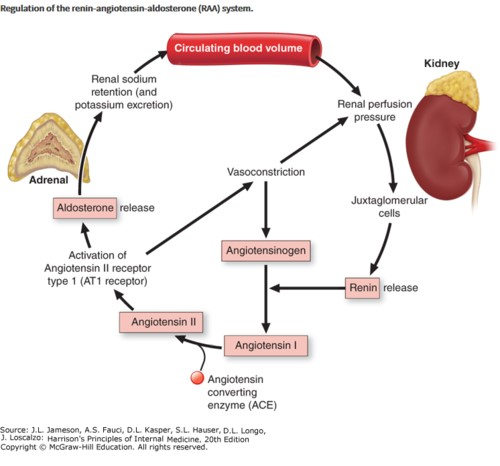
Numerous RAS (renin-angiotensin system) components have been found in a variety of organs, including adipose tissue, suggesting that this system may possibly be involved in the control of local tissue functions. Furthermore, there is evidence for a function for the renal RAS in the etiology of diabetic nephropathy, and the intrarenal RAS may work independently of the systemic renin-angiotensin in diabetic individuals. Angiotensinogen, Angiotensin II, Rennin, and Aldosterone plasma levels appear to be unaffected by diabetes in animal models, according to studies. However, early on, the local renal RAS is activated, increasing the angiotensin II production in the area. Others have discovered that plasma renin activity and serum aldosterone levels are lowered in type 1 and type 2 human diabetes as well as animal diabetes. Diabetes-related hyporeninemic hypoaldosteronism has been linked to a number of illnesses, including hypertension, nephropathy, neuropathy with diminished sympathetic nerve activity, and angiopathy. Researchers have observed enhanced renal renin system activity in animals and people with type 1 and type 2 diabetes, and they have hypothesized that this may be due to autonomous and unsuppressed local RAAS function. It’s probable that the variations in the RAS function identified in diabetes represent several stages in the development of the diabetic condition [23].
Angiotensin II binding to its receptor in the glomerulus and angiotensin receptor 1 (AT1) density are both diminished at the receptor level in animal diabetes. The phenomenon is reversed by insulin therapy. On the other hand, diabetes increases myocardial AT receptors, and this alteration is less sensitive to insulin therapy. These findings imply that in diabetes, the AT receptors are differentially regulated in the kidney and heart. Finally, type 2 diabetes patients have an unaffected aldosterone response to angiotensin II at the adrenal level. Together, it appears that diabetes may cause an early stage of RAAS hyperactivation and a later stage of hypo-reninemic hypoaldosteronism. Diabetics may exhibit enhanced renal RAS activity, which could account for their positive response to angiotensin-converting enzyme inhibitor therapy. Thyroid Gland: In stable, under control diabetic patients, plasma thyroid hormone levels are typically normal, and hormone levels are similar in people with type 1 or type 2 diabetes. Diabetes, however, may also affect the peripheral hypothalamic-pituitary-thyroid axis in several ways. Uncontrolled diabetes mellitus affects the peripheral T3 and reverse T3 (rT3) levs, reflecting a problem with T4 peripheral mono deiodination, similar to many other non-thyroidal diseases. The levels of T3 and glycosylated hemoglobin are negatively correlated, and individuals with diabetic ketoacidosis exhibit the greatest fall in T3 levels. After improving glycemic control, the aberrant thyroid hormone levels typically return to normal. Conflicting results are seen when type 1 and type 2 diabetes individuals’ TSH secretion is evaluated. TSH levels have been described as rising, being normal, or falling. Diabetes has no impact on the metabolism of exogenous TSH, despite similar inconsistent findings regarding the TSH response to TRH stimulation in diabetic individuals.
Diabetes of both types was found to blunt the nocturnal TSH peak, a sensitive indicator of the hypothalamic-pituitary- thyroid axis’s central control. Better glycemic management can correct these aberrations in TSH secretion. Because of this, acute hyperglycemia in people with uncontrolled diabetes and in animals with diabetes may result in thyroid abnormalities that resemble secondary or tertiary hypothyroidism. Parathyroid Gland: The impact of diabetes on PTH levels varies and may be time-dependent. PTH levels in rats with short-term diabetes rise, whereas PTH levels in rats with long-term diabetes decline. Others discovered normal PTH levels but a reduced ability of 25(OH) Vit D to convert to 1,25(OH) 2 Vit D in response to PTH. There are conflicting reports on PTH levels in diabetic people. PTH levels in both type 1 and type 2 diabetics were found to be within the normal range by some researchers, whereas type 1 diabetes patients were shown to have lower PTH levels (Figure 11).
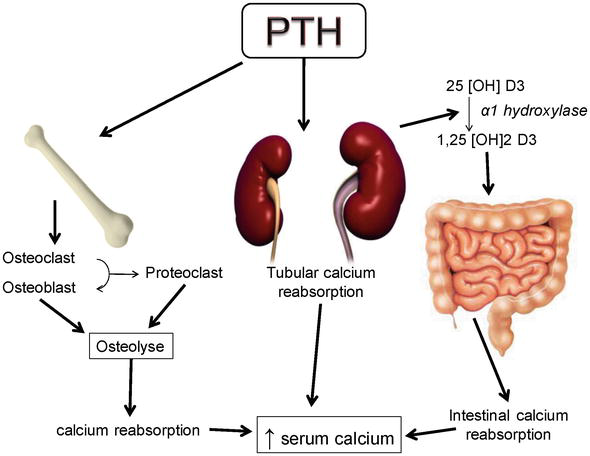
PTH levels are lower in people with type 1 diabetes than they are in people with type 2 diabetes. PTH may boost the synthesis of insulin, but it may also lessen the hormone’s peripheral impact. It’s interesting to note that primary hyperparathyroidism and the prevalence of diabetes mellitus go hand in hand. Around 8% of people with primary hyperparathyroidism have diabetes mellitus, while only 1% of people with type 1 or type 2 diabetes have primary hyperparathyroidism. The higher prevalence of diabetes in these patients may be explained by the higher incidence of insulin resistance associated with primary hyperparathyroidism. On the other hand, low 1,25(OH)2 Vit D may be to blame for the continuous stimulation of the parathyroid glands, which results in an increased incidence of primary hyperparathyroidism in diabetic individuals.
Osteopenia and bone fractures are more common in those with type 1 diabetes. In people with microalbuminuria, the low-turnover osteopenia found in type 1 diabetes is more severe. The causes of osteopenia are multifaceted and can include hypercalciuria, low 1,25(OH)2 vitamin D levels, low calcium levels, and reduced osteoid formation. The data in those with type 2 diabetes are less consistent [24].
Adrenal Androgens: Male type 2 diabetic patients had lower serum levels of dehydroepiandrosterone (DHEA) and dehydroepiandrosterone sulfate (DHEA-S). It has been demonstrated that a higher incidence of atherosclerosis in men is correlated with lower serum levels of DHEA and DHEA-S. Therefore, the drop in DHEA and DHEA-S levels in type 2 diabetics may be a factor in this population’s higher prevalence of ischemic heart disease. DHEA levels could further decline due to poor glycemic management.
Serum insulin levels and DHEA and DHEA-S levels are inversely correlated. Regardless of glycemic level, normal men and women’s serum DHEA-S levels significantly decreased after experimentally produced hyperinsulinemia. Others have noted that hyperglycemia causes a drop in both DHEA and DHEA-S in male patients with type 2 diabetes or impaired glucose tolerance (IGT), and that this decline is unrelated to blood insulin levels. There is no difference between the amounts of DHEA-S and androstenedione seen in patients with type 1 diabetes and those seen in their nondiabetic siblings, in contrast to observations in type 2 diabetes.
DHEAS levels are normal or elevated in type 2 diabetic females (Table 1).
| Effect on Glucose Metabolism | |
|---|---|
| Anterior Pituitary | Hypersecretory Conditions |
| GH Hypersecretion | Insulin Resistance Diabetes Mellitus |
| ACTH Hypersecretion | Impaired Glucose Tolerance Diabetes Mellitus |
| Prolactin Hypersecretion | Prolactinoma - Higher FPG in Small Studies Physiological Hyperprolactinaemia - Insulin Resistance? |
| TSH Hypersecretion | Insulin Resistance, Worsening of Diabetes |
| Anterior Pituitary | Hyposcretory Conditions |
| Panhypopituitarism | Hypoglycaemia |
| GH Deficiency | Children - Insulin Sensitivity, Fasting Hypoglycaemia Adults - Insulin Resistance |
Table 1: Disease states effecting on glucose metabolism.
Diabetes Mellitus Types
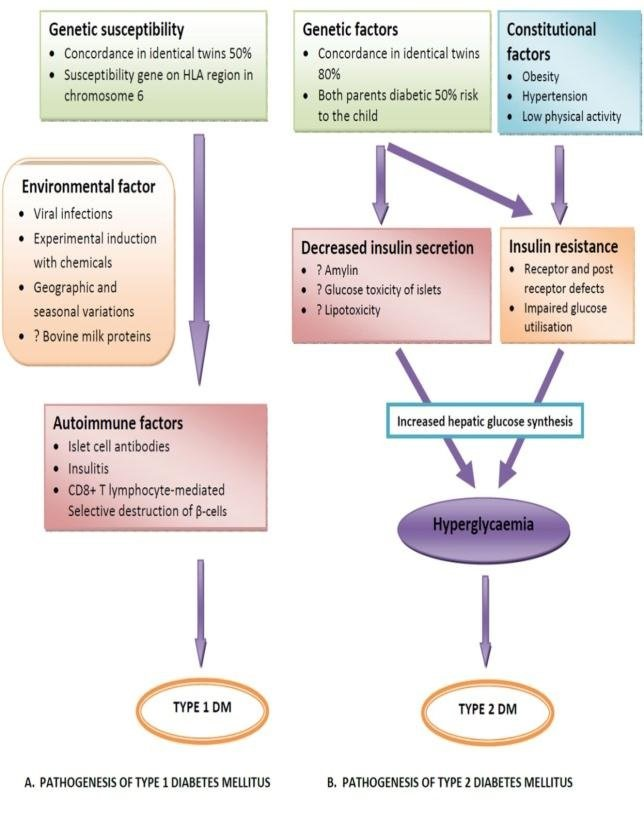
Type 1 Diabetes Mellitus: The pancreatic B-cells are destroyed by the immune system in type1 DM. According to current theories, type 1 diabetes in people who are genetically vulnerable is brought on by exposure to an environmental trigger. However, less than 10% of the genetically predisposed people have B-cell autoimmunity, and less than 1% advance to type 1 diabetes. While 90% of people had B-cell autoimmunity, including ICAs, at the time of diagnosis. A genetically predisposed person needs to be exposed to a trigger that starts the autoimmune process and kills pancreatic beta-cells in order for type 1 DM to manifest. However, it is unclear what exactly the motivating reasons are. Early exposure to cow’s milk, not nursing, gut bacteria (i.e., intestinal microbiome), and specific viruses (such as enterovirus and rotavirus) have all been identified as triggers.
Although vitamin D insufficiency is more common in patients who develop type 1 diabetes, it is uncertain if this link or causality exists. • Macrophages and T lymphocytes with circulating autoantibodies to different -cell antigens mediate the autoimmune process.
Islet cell autoantibodies (ICAs) are the antibodies most often linked to type 1 diabetes. There is a prolonged preclinical stage during which autoimmune markers can be found in many people who develop type 1 DM. Cell autoimmunity may exist for up to 13 years before type 1 diabetes is diagnosed.
- In some people, autoimmune disease remits, whereas in others, it develops to complete –cell failure.
- When 60% to 90% of the -cells are destroyed, hyperglycemia develops.
- The pancreatic beta-cell also secretes another hormone called amylin along with insulin.
- Due to the loss of -cells, patients with type 1 DM also lack amylin.
- Amylin slows stomach emptying, reduces improper glucagon secretion, and promotes central satiety.
Type 2 Diabetes Mellitus: Type 2 DM, also known as non-insulin-dependent diabetes or adult-onset diabetes, is brought on by -cell malfunction and a certain amount of insulin resistance. There is a steady loss of cells throughout time. Overweight or obese people with type 2 diabetes predominate. Insulin resistance is largely attributed to abdominal obesity. Since type 2 DM has a clear inheritance pattern, genetics are crucial in its development. The majority of genetic changes linked to type 2 diabetes (T2DM) seem to have an impact on the growth and operation of -cells, the sensitivity of cells to insulin action, or the onset of obesity [25].
Type 2 diabetes mellitus is likely polygenetic, with multiple genetic abnormalities contributing to its pathophysiology and a wide variety of combinations of derangements influencing its occurrence in various populations. High blood pressure and dyslipidemia, which is characterized by high serum triglycerides and low HDL cholesterol levels, are highly common coexisting disorders in people with type 2 diabetes. Additionally, typical is increased serum plasminogen activator inhibitor-1 (PAI-1), which adds to a hypercoagulable state. The majority of people who acquire type 2 DM have several abnormalities that affect how plasma glucose is regulated, including:
Impaired insulin secretion, insufficient incretin hormones, muscle, liver, and adipocyte insulin resistance, excessive glucagon secretion, increased hepatic glucose production, upregulated sodium-glucose cotransporter in the kidney, systemic inflammation, and eight other factors are all contributors to insulin resistance. Patients with type 2 DM may even have a paradoxical spike in glucagon levels because they are unable to inhibit glucagon in response to a meal.
Several Elements Influence:
- Lack of or resistance to GLP-1
- Insulin sensitivity
- a lack of insulin.
Increases in GLP-1 and insulin after meals would stop the release of glucagon. As a result, the liver produces an excessive amount of glucose as a result of hyperglucagonemia. 90% of the filtered glucose is reabsorbed in the kidney by the proximal renal tubular cells that contain the SGLT- 2 transporter. Sodium glucose cotransporter-1 (SGLT-1) reabsorbs 10%. The renal threshold is raised in diabetic patients from 210 to 240 mg/dL before glycosuria starts. The enhanced expression of SGLT-2 receptors is the cause of the glucose’s more aggressive reabsorption.
Hyperglycemia is further exacerbated by an increase in the kidney’s reabsorption of glucose.
Gestational Diabetes: GDM develops during pregnancy. Increased insulin resistance is brought on by hormonal changes during pregnancy, and GDM may arise if the mother is unable to raise her insulin secretion to enough make up for this and maintain normoglycemia. GDM-affected women are more likely to later acquire type 2 DM. The majority of the time, glucose intolerance develops at the start of the third trimester. Assessment and intervention ought to start at the very first prenatal appointment. Because treatment will lower prenatal morbidity and mortality, detection is crucial. Other Types of Diabetes: Young people with maturity- onset diabetes (MODY) have reduced insulin production in response to a glucose stimulation and little to no insulin resistance. Mild hyperglycemia usually appears in patients at a young age, but diagnosis is frequently delayed. At least six distinct mutations have been found to date, and the condition is inherited in an autosomal-dominant form. Most often, MODY 2 and 3 are used. A few families have been shown to produce mutant insulin molecules, which also causes aberrant glucose intolerance. The clinical syndrome of type A insulin resistance includes acanthosis Nigerians, civilization of females, polycystic ovaries, and hyperinsulinemia. Antibodies to the insulin receptor may prevent insulin from attaching. This condition is known as type B insulin resistance.
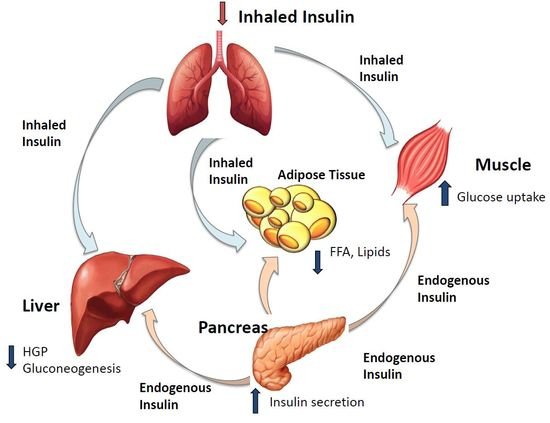
The Pancreas and its Hormones
Location: The pancreas is located in the abdomen, behind the stomach, and serves two distinctly different functions.
Role: First, it acts as an exocrine organ, because the majority of pancreatic cells produce various digestive enzymes that are secreted into the gut and which are essential for the effective digestion of food. Second, the pancreas serves as an endocrine organ, because certain cell clusters (i.e., the Islets of Langerhans) produce two hormones—insulin and glucagon—that are released into the blood and play pivotal roles in blood glucose regulation. Insulin: Insulin is produced in the beta cells of the Islets of Langerhans. Its primary purpose is to lower blood glucose levels; in fact, insulin is the only blood sugar-lowering hormone in the body. To this end, insulin promotes the formation of storage forms of energy (e.g., glycogen, proteins, and lipids) and suppresses the breakdown of those stored nutrients.
Accordingly, the target organs of insulin are primarily those that are specialized for energy storage, such as the liver, muscles, and adipose tissue. Specifically, insulin has the following metabolic effects.
Promotes glucose uptake into cells and its conversion into glycogen, stimulates the breakdown of glucose, and inhibits gluconeogenesis. Stimulates the transport of amino acids into cells and protein synthesis in muscle cells, thereby lowering the levels of amino acids available for gluconeogenesis in the liver. Increases fat synthesis in the liver and adipose tissue, thereby lowering the levels of glycerol, which also can serve as a starting material for gluconeogenesis.
The release of insulin is controlled by various factors, including blood glucose levels; other islet hormones (e.g., glucagon); and, indirectly, other hormones that alter blood glucose levels (e.g.: GH, glucocorticoids, and thyroid hormone). Dysfunction of Insulin Causes DM: Type 2 diabetes mellitus is characterized by 4 major metabolic abnormalities: obesity, impaired insulin action, insulin secretory dysfunction, and increased endogenous glucose output (EGO). Although there is substantial evidence that the first 3 of these abnormalities are present in most individuals before the onset of diabetes, the sequence with which they develop and their relative contributions to the progression from normal glucose tolerance (NGT) to impaired glucose tolerance (IGT), and ultimately to type 2 diabetes, remain unknown in the absence of a detailed longitudinal study [26].
In cross-sectional studies, subjects with IGT were on average more obese and more insulin-resistant than those with NGT. Basal EGO, largely reflecting hepatic glucose production, was not increased (Figure 12).
Whether insulin secretion is impaired in individuals with IGT is controversial. Some studies have found a lower early insulin secretory response (occurring within minutes of an intravenous or oral glucose load) in individuals with IGT compared with those with NGT. In recent years, several prospective studies, in which nondiabetic individuals are metabolically characterized single occasion and then followed for several years to determine who develops diabetes, have helped to identify metabolic abnormalities that predispose to diabetes. A low early insulin response predicted diabetes in most but not all studies. The defects in both insulin action and insulin secretion predispose some individuals with NGT to diabetes, but they give little information about the time course with which these abnormalities change as glucose tolerance worsens.
Keywords • Insulin-stimulated glucose disposal (M)
- Maximally insulin-stimulated glucose disposal (M-high)
- Minimum insulin-stimulated glucose disposal (M-low)
- Acute insulin secretory response (AIR) Insulin Action: The transition from NGT to diabetes was accompanied by a progressive deterioration of insulin action, as indicated by a decrease in M-high. This decrease was evident during the transition from NGT to IGT, indicating that insulin action declines at an early stage of the disease. The decreased glucose uptake was almost entirely due to a decrease in nonoxidative glucose disposal, suggesting that the impairment of insulin action during the development of diabetes is primarily caused by a defect in glucose storage. Subjects with type 2 diabetes are resistant to the stimulating effect of insulin on skeletal muscle glycogenesis. The decrease in glucose uptake at physiological insulin levels (M-low) in the progressors was smaller than the decrease in M-high and did not differ from that observed in the nonprogressors (Figure 13).
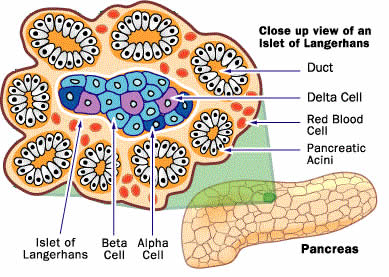
Insulin Secretion: The decrease in the AIR to glucose during the transition from NGT to IGT unequivocally demonstrates that defects in insulin secretion occur at an early stage during the development of type 2 diabetes. Islets of Langerhans • Introduction: The islands or (more commonly) islets of Langerhans, first described by their namesake- Paul Langerhans- in 1969, are islands of mixed populations of endocrine cells that are scattered in the parenchyma of the pancreas. Islets of Langerhans have been much studied in the context of diabetes due to the hormones produced and secreted from the cells which form these micro-organs, which are involved in the regulation of glucose homeostasis [27].
Islets of Langerhans are islands of endocrine cells scattered throughout the pancreas. A number of new studies have pointed to the potential for conversion of non-ß islet cells in to insulin-producing B-cells to replenish B-cell mass as a means to treat diabetes.
Islet Distribution in the Pancreas • Structure, Size, Location: Human pancreas contains on average 3.2 million islets, with a mean islet diameter of 108.92 um (+6.27 um), and a mean islet volume of 0.00069 uL (+0.00011 uL). The majority of islets (~66%) have a surface area of between 1000 and 10,000 um2. Approximately 24% and 9% of islets had a surface area of more than 100,000 um2 and less than 100 um2, respectively (Figure 14).
The islets appear to have a threshold surface area of about 100,000 um2, where “islets” that have apparently larger surface area than the threshold were made up of clusters of islets. The smaller islets tended to cluster around blood vessels. The islet surface area and distribution were averaged over pancreas section area, there is a uniform scattering of islets in the body of the pancreas, with the islets adopting an overall spherical structure although the differences may be due to differences in the methodologies used for estimating B-cell mass (Figure 15).
• Types of Islet of Langerhans:
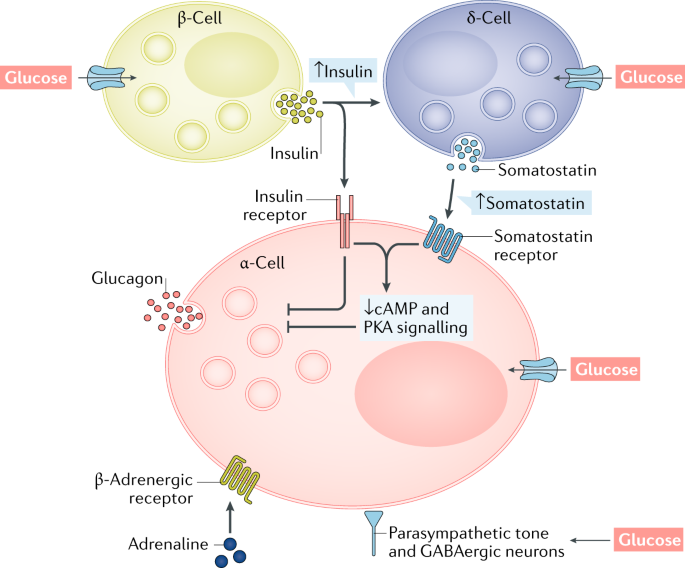
- a-Cell:
- Function: The a-Cell of the pancreas are responsible for producing and secreting the hormone glucagon. Glucagon plays several important roles in regulating blood glucose levels:
- Stimulating Glycogenolysis: Glucagon triggers the breakdown of glycogen into glucose in the liver, increasing blood sugar levels. This is important when blood sugar levels are too low, such as between meals or during physical activity.
- Promoting Gluconeogenesis: Glucagon promotes gluconeogenesis, which is the synthesis of glucose from non-carbohydrate sources like amino acids and glycerol. This process also helps raise blood sugar levels.
- Inhibiting Glycolysis: It inhibits glycolysis, the breakdown of glucose in cells, helping to conserve glucose for other cells that require it.
- B-Cell:
- Function: The primary function of beta cells is to synthesize, store, and release insulin in response to changes in blood glucose levels. Insulin is a hormone that helps regulate blood sugar (glucose) by promoting the uptake of glucose into cells, particularly muscle and fat cells, and by inhibiting the release of glucose from the liver.
- Insulin Production: Beta cells are responsible for producing and secreting insulin in response to elevated blood glucose levels, such as after a meal. This release is a tightly regulated process to maintain blood glucose within a narrow range.
Glucose Sensing: Beta cells have glucose-sensing receptors that enable them to detect changes in blood glucose concentrations. When blood sugar rises, beta cells respond by releasing insulin to lower it.
Insulin Secretion: Beta cells release insulin through a process that involves vesicles containing pre-formed insulin molecules. When stimulated by increased glucose, these vesicles fuse with the cell membrane, allowing insulin the bloodstream.
Regulation: Various factors, including hormones, neural signals, and nutrients, can influence the secretion of insulin from beta cells. For example, glucagon, another hormone produced in the pancreas, can stimulate beta cells to release insulin when blood glucose is too high [28].
Beta Cell Dysfunction in Diabetes:
Dysfunction:
Dysfunction or destruction of beta cells is a central feature of type 1 diabetes, where the immune system attacks and destroys these cells. In type 2 diabetes, beta cells may lose their ability to secrete insulin effectively in response to high blood sugar, contributing to the disease. The balance between insulin (produced by beta cells) and glucagon (produced by alpha cells) is crucial in regulating blood glucose levels to keep them within a narrow range. When blood sugar is too high, insulin is released to lower it, while when it’s too low, glucagon is released to raise it. This regulation is essential for maintaining overall metabolic health.
Beta Cell Proliferation:
Mechanisms: During neonatal life, the endocrine pancreas undergoes substantial remodelling, which involves significant apoptosis, replication, and neogenesis of islet cells. Islet mass grows into adulthood to match increased hormonal demand. In contrast, there is little change in islet mass in adults except in response to physiological and/or pathological changes such as pregnancy and obesity.
By GSIS, beta cells tightly regulate systemic glycaemia within narrow range. Insufficient functional beta cell mass is the causal factor of type 1 and a major contributor to type 2 diabetes, emphasizing the importance of understanding cell dynamics. Signals controlling beta cell number may therefore act by modulating the survival and/or proliferation of differentiated beta cells.
Uncontrolled beta cell proliferation will increase beta cell numbers and enhance the functional capacity. However, hypoglycaemia could be a consequence of increased, uncontrolled beta cell proliferation due to an excess of beta cells, and the resultant over secretion of insulin. Further, increased beta cell numbers will occur at the expense of other islet cell types that secrete hormones antagonistic to insulin to also facilitate glucose homeostasis. Disrupting the islet cell population will limit the ability for maintaining glucose homeostasis. Apoptosis may also be potentiated to regulate uncontrolled proliferation. The balance between sustaining proliferation and evading apoptosis is critical for the initial beta cell compensatory response to normalize glycaemia. This delicate shift to either physiological process has to adapt to metabolic demand. If this balance is disrupted to favour either process it would exacerbate the diabetogenic state.
Beta Cell Physiology and Integrity:
Insulin Production: Beta cells are responsible for producing and releasing insulin, a hormone that regulates blood sugar. When blood glucose levels rise (e.g., after a meal), beta cells release insulin to help cells take in and use glucose for energy.
Glucose Sensing: Beta cells have glucose-sensing mechanisms that allow them to detect changes in blood glucose levels. Glucose enters beta cells through glucose transporters and triggers insulin release.
Insulin Secretion: Insulin secretion is tightly regulated. It involves a complex interplay of signalling pathways and molecules, including ATP-sensitive potassium channels, calcium ions, and various enzymes.
Integrity and Function: Maintaining the integrity and function of beta cells is crucial for overall health. Factors like chronic high blood sugar, inflammation, and autoimmune responses can damage or destroy beta cells, leading to conditions like diabetes.
Beta Cell Mass: The number and health of beta cells in the pancreas, known as beta cell mass, influence an individual’s ability to regulate blood sugar. Loss of beta cell mass can result in insulin deficiency and diabetes.
Diabetes: Type 1 diabetes is characterized by an autoimmune attack on beta cells, leading to their destruction and insulin deficiency. In Type 2 diabetes, beta cells often become less responsive to glucose, and their mass may decrease time.
Protection and Research: Efforts to protect and regenerate beta cells are a focus of diabetes research. This includes studying factors that promote beta cell survival and developing potential treatments like beta cell transplantation or regenerative therapies.
Maintaining the health and function of beta cells for preventing and managing diabetes and related metabolic disorders.
Beta Cell Demise and Dysfunction: Beta cell demise and dysfunction are central issues in the context of diabetes mellitus, particularly in type 1 and type 2 diabetes.
Several factors contribute to beta cell demise and dysfunction:
Autoimmune Response: In type 1 diabetes, an
autoimmune response causes the immune system to target and destroy beta cells. Maintaining the health and function of beta cells is essential for preventing and managing diabetes and related metabolic disorders.
Beta Cell Demise and Dysfunction: Beta cell demise and dysfunction are central issues in the context of diabetes mellitus, particularly in type 1 and type 2 diabetes [29].
Several factors contribute to beta cell demise and dysfunction:
Autoimmune Response: In type 1 diabetes, an autoimmune response causes the immune system to target and destroy beta cells.
Insulin Resistance: In type 2 diabetes, chronic insulin resistance places increased demand on beta cells, which can eventually exhaust them.
Genetic Factors: Genetic predisposition plays a role in both types of diabetes, influencing susceptibility to beta cell dysfunction.
Environmental Factors: Lifestyle factors like poor diet, lack of exercise, and obesity can contribute to beta cell stress and dysfunction.
Oxidative Stress and Inflammation: These factors can also damage beta cells and reduce their ability to produce insulin. Addressing beta cell demise and dysfunction is a key focus in diabetes research and treatment. Therapies aim to preserve existing beta cell function, stimulate their regeneration, or replace them with transplantation or other innovative approaches. Additionally, lifestyle changes, such as maintaining a healthy weight and managing blood sugar levels, can help protect beta cell function in type 2 diabetes.
Saturated Fat and FFA Trigger Beta Cell:
Dysfunction and Insulin Resistance: Saturated fat and elevated levels of free fatty acids (FFA) have been associated with beta cell dysfunction and insulin resistance. Consuming a diet high in saturated fats can lead to an accumulation of FFA in the bloodstream, which can impair the function of insulin-producing beta cells in the pancreas. This, in turn, can reduce insulin secretion and lead to insulin resistance in target tissues like muscles and liver. It’s important to maintain a balanced diet to support overall metabolic health and reduce the risk of these issues.
The reasons saturated fat and elevated levels of free fatty acids (FFA) can trigger beta cell dysfunction and insulin resistance is multifaceted:
Lipotoxicity: Saturated fats, when consumed in excess, can lead to the accumulation of lipids within beta cells. This intracellular lipid build up can impair beta cell function and insulin secretion, a phenomenon known as lipotoxicity.
Oxidative Stress: High saturated fat intake can promote oxidative stress within beta cells. Oxidative stress can damage cellular components and impair insulin production and signalling.
Inflammation: Saturated fats can induce low-grade inflammation in the body. Chronic inflammation is closely linked to insulin resistance and beta cell dysfunction. Mitochondrial Dysfunction, Saturated fats may disrupt mitochondrial function in beta cells, which is essential for insulin production. Impaired mitochondrial function can lead to reduced insulin secretion.
FFA Interference: Elevated levels of free fatty acids, often associated with high saturated fat intake, can interfere with insulin signalling in peripheral tissues (muscles and liver). This interference can lead to insulin resistance, where these tissues do not respond effectively to insulin.
Endoplasmic Reticulum Stress: Saturated fats and high FFAs can induce endoplasmic reticulum (ER) stress in beta cells. ER stress disrupts cellular processes and can impair insulin production and secretion.
Beta Cell Apoptosis: Prolonged exposure to high levels of saturated fats and FFAs may lead to beta cell apoptosis (cell death). This results in a decreased number of functional beta cells, which further impairs insulin secretion.
P-Cell: Somatostatin-releasing cells, also known as delta cells, are a type of endocrine cell found in the pancreas. These cells are primarily located in the islets of Langerhans, which are clusters of cells within the pancreas responsible for regulating blood sugar levels. Somatostatin is a hormone produced and secreted by these delta cells.
The primary function of somatostatin is to inhibit the secretion of other hormones, including insulin and glucagon, both of which play crucial roles in blood glucose regulation. By releasing somatostatin, delta cells help modulate the activity of neighbouring alpha and beta cells in the islets of Langerhans, thus contributing to the fine-tuning of blood sugar levels in the body. This hormone also has various other functions in the body, such as inhibiting the release of growth hormone, gastrin, and several gastrointestinal hormones [30].
PP Cell: Pancreatic polypeptide containing cells, also called PP cells or F-cells make up 1-2% of the islet cell population. PP cells are more concentrated in the head of the pancreas where the cells are found to occupy the outer mantle of rodent islets or lining the capillaries in human islets. Post-prandial pancreatic polypeptide release is regulated by vagal and enteric nervous input and is responsive to arginine but not glucose stimulation.
Functions: Pancreatic polypeptide has been shown to be an inhibitor of glucagon release at low glucose. The major function of PP appears to be that of a satiety hormone.
Inhibiting Pancreatic Secretion: Pancreatic
polypeptide can reduce the secretion of pancreatic enzymes, which important for the digestion of fats and proteins. This helps slow down the digestive process, allowing the body to absorb nutrients more effectively.
Reducing Appetite: It plays a role in regulating appetite and food intake. After a meal, the hormone is released to signal a sense of fullness, which can help control food consumption.
Regulating Gastrointestinal Motility: Pancreatic polypeptide can affect the movement of food through the digestive tract, helping to regulate the rate of digestion and absorption of nutrients.
Promoting Liver Glycogen Synthesis: It can stimulate the liver to store glucose in the form of glycogen, which helps maintain blood sugar levels.
Ghrelin-Positive and Other Islet Cell Types: A further three types of islet cells have been described in the literature. These cells contain ghrelin, serotonin (enterochromaffin cells), gastrin (G-cells) and small granules of unknown content (P/D1-cells). Of these the ghrelin positive cells have recently attracted the most interest. Ghrelin-positive cells are mainly found in the gut. Ghrelin-positive cells are also found in the islet, accounting for circa 10% and 1% of islet cell content in foetal and adult islets, respectively. It has been suggested that ghrelin positive cells in the islets are in fact the P/D1 cells, which were described as containing an unknown hormone, as the two cell types share a lot of ultrastructural and distribution similarities.
Ghrelin is increased in fasting plasma ghrelin content has a reciprocal relationship with plasma insulin content and has been shown to be an inhibitor of insulin secretion in human.
Function: Ghrelin may also be a regulator of glucagon, PP and somatostatin release.
Glucagon: The second blood-sugar–regulating pancreatic hormone is glucagon, which is produced in the alpha cells of the Islets of Langerhans. Glucagon increases blood glucose levels; accordingly, its main actions generally, are opposite to those of insulin. For example, glucagon increases glycogen breakdown and gluconeogenesis in the liver as well as the breakdown of lipids and proteins. The release of glucagon is regulated by many of the same factors as is insulin’s release, but sometimes with the opposite effect. Thus, an increase in blood glucose levels stimulates insulin release but inhibits glucagon release. A finely tuned balance between the activities of insulin and glucagon is essential for maintaining blood sugar levels. Accordingly, disturbances of that balance, such as an insulin deficiency or an inability of the body to respond adequately to insulin, result in serious disorders, such as diabetes mellitus [31].
The Role of Glucagon: The pancreas releases glucagon when glucose levels fall too low. Glucagon causes the liver to converted glycogen into glucose, which is released to the bloodstream. High Blood glucose stimulate the release of Insulin allows glucose to be taken up and used by insulin independent tissues such as muscle cell. Glucagon and insulin work together automatically as a negative feedback system to keep blood glucose levels. People with type 2 diabetes have excess glucagon secretion, which is a contribution for chronic hyperglycemia of type 2 diabetes. The amazing balance of these two opposing hormones of glucagon and insulin is maintained by another pancreatic hormone called somatostatin, created in the delta cells.
Regulation of Glucagon Action
Glucagon Action Regulation: Clearly, restoring glucose homeostasis has benefited from the use of insulin replacement treatment. However, it is merely a portion of the overall answer. The crucial connection between glucagon and insulin has proposed alternative therapeutic horizons. The activities of glucagon are excessive due to inadequate insulin levels and increased glucagon levels in the portal vein, which results in an endogenous and superfluous supply of glucose in the fed state. The reduction of postprandial glucagon.
Regulation of Glucagon Secretion: In order to prevent hypoglycemia, the hormone glucagon stimulates the hepatic synthesis of glucose. It is generally known that the release of glucagon in response to hypoglycemia is mediated by autonomic, endocrine, and paracrine systems, and that the alpha cells likely directly sense blood glucose levels.
Although the precise molecular processes of glucagon production from alpha cells are not well understood, they involve potassium-adenosine triphosphate (K ATP) channels and voltage-gated calcium channels. The beta cell secretory products insulin, zinc, and gamma-aminobutyric acid (GABA), which are released in response to meals and rising blood sugar levels, block glucagon secretion during hyperglycemia in normal physiology. When blood sugar levels drop, beta cells produce fewer of these substances, eliminating their inhibitory influence on alpha cells. Increased glucagon secretion in response to hypoglycemia is the end result of this mechanism, often known as the “switch-off” hypothesis. Somatostatin, which is produced by the pancreatic islets’ delta cells, and GLP-1 also prevent the release of glucagon [32].
A significant part of the release of glucagon is also played by autonomic activity in response to hypoglycemia.
It has been demonstrated that circulating adrenaline, as well as sympathetic and parasympathetic stimulation, all increase glucagon release. The autonomic nervous system is subsequently activated to prompt glucagon release when glucose sensors in the ventromedial hypothalamus detect hypoglycemia. A postprandial dip in glucose that could happen with unopposed insulin secretion is likely avoided by increased glucagon production in response to a protein- rich meal. • Action and Effect: Glucagon is released by the alpha cells and travels from the pancreatic veins to the portal venous system. Hepatocytes contain glucagon receptors, and the kidney, heart, digestive system, adipose tissue, brain, and spleen have all been found to contain glucagon binding sites. Following glucagon binding to its receptor in the hepatocytes, linked G proteins are activated to initiate downstream signaling. Adenylate cyclase is activated by the GS- alpha subunit, which raises the amount of cyclic AMP (cAMP) found inside of cells. Hepatic glucose production (HGP) and plasma blood glucose levels rise as a consequence.
• Pathophysiology of Diabetes: The pathophysiology of diabetes is becoming increasingly clear to us. The loss of pancreatic cells caused by type 1 diabetes has been described as an autoimmune-mediated process. Amylin, the other co-secreted and co-located -cell hormone, is likewise deficient as a result of the low insulin and decreasing availability of insulin, amylin, and GLP- 163. Both type 1 and type 2 diabetes frequently cause abnormal stomach emptying. Postprandial glucose concentrations are significantly influenced by the velocity of stomach emptying. Accelerated stomach emptying causes insulin administration to be poorly timed with the presentation of meal-derived glucose to the circulation. In people with diabetes, postprandial hyperglycemia is made worse by the absence or delayed production of insulin. By delaying the passage of nutrients from the stomach to the small intestine, both amylin and GLP-1 control gastric emptying (Figure 16).
The absence of an initial insulin response to a meal is a sign that postprandial -cell action has become abnormal early in type 2 diabetes. The clinical picture of hyperglycemia in diabetes is influenced by peripheral insulin resistance, increasing –cell failure, the metabolic abnormalities associated with IDDM are caused by a lack of insulin secretion, which is caused by the autoimmune death of pancreatic beta- cells. Patients with IDDM also have abnormal pancreatic -cell function and increased glucagon secretion in addition to the lack of insulin secretion. Normally, hyperglycemia causes glucagon secretion to decrease; however, in patients with IDDM, glucagon secretion is not inhibited by hyperglycemia.
- Pathophysiology of Type 1 Diabetes Mellitus:
- Type 1 Diabetes Mellitus (T1DM): Type 1 Diabetes Mellitus (T1DM), often referred to as just Type 1 Diabetes, is a chronic medical condition characterized by the body’s inability to produce insulin. Insulin is a hormone produced by the pancreas that is essential for regulating blood sugar (glucose) levels. In T1DM, the immune system mistakenly attacks and destroys the insulin-producing beta cells in the pancreas, leading to a deficiency of insulin [33]. Type 1 diabetes is an autoimmune disease that primarily affects the beta cells in the pancreas.
- Autoimmune Destruction of Beta Cells: In Type 1 diabetes, the body’s immune system mistakenly targets and destroys the insulin-producing beta cells in the pancreas. This autoimmune process is thought to be triggered by genetic and environmental factors, although the exact cause is not fully understood.
- Insulin Deficiency: As beta cell destruction progresses, the pancreas produces very little to no insulin. This results in an absolute insulin deficiency, which leads to elevated blood glucose levels because insulin is essential for the uptake of glucose into cells.
- Hyperglycemia: With inadequate insulin, glucose cannot enter cells to provide energy, causing high blood sugar levels (hyperglycemia). The absence of insulin also triggers the release of counter regulatory hormones (e.g., glucagon and cortisol), exacerbating hyperglycemia (Figure 17).
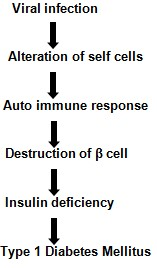
- Pathogenesis of type 1 Diabetes Mellitus: A chronic autoimmune condition called type 1 diabetes mellitus is linked to the selective eradication of insulin- producing pancreatic -cells. The last stage of the process that destroys -cells and results in type 1 diabetes mellitus is when the disease manifests clinically.
- The presence of auxiliary and immune-competent cells in infiltrating pancreatic islets;
- The class II (immune response) genes of the major histocompatibility complex (MHC; human leucocyte antigens HLA) are associated with illness vulnerability;
- The presence of autoantibodies specific for islet cells;
- Modifications in CD4+ T cell compartment immuno- regulation, in particular;
- The role of monokines and TH1 cells, which produce interleukins, in the illness process;
- Immunotherapy response.
- The affected people or their family members frequently develop other autoimmune disorders that are unique to specific organs [34].
The metabolic abnormalities carried on by insulin deficiency is made worse by the consequent, abnormally high glucagon levels. The most obvious manifestation of this metabolic disruption is the quick onset of diabetic ketoacidosis in IDDM patients in the absence of insulin therapy. Lack of insulin causes unchecked lipolysis and higher plasma levels of free fatty acids, which inhibit the metabolism of glucose in peripheral tissues such skeletal muscle. This affects how well the body uses glucose, and insulin deprivation also causes a number of genes to express less, including those for the GLUT4 class of glucose transporters in adipose tissue and glucokinase in the liver, which are both required for the target tissues to respond to insulin appropriately. Impaired glucose, lipid, and protein metabolism are the main metabolic abnormalities caused by insulin insufficiency in IDDM. These abnormalities are detailed in more depth as follows:
- Effect on Glucose Metabolism: Hepatic glucose production increases when IDDM is uncontrolled. Hepatic gluconeogenesis is then used to create glucose after first mobilizing liver glycogen reserves. Lack of insulin also affects how well glucose is used by non- hepatic tissues. Insulin promotes glucose absorption, especially in adipose tissue and skeletal muscle. This is achieved via the migration of glucose transporter proteins to the plasma membrane of these tissues, which is mediated by insulin. Reduced peripheral tissue uptake of glucose results in a slower rate of glucose metabolism. In addition, insulin controls the amount of hepatic glucokinase. As a result, greater blood delivery results from a decreased rate of glucose- phosphorylation in hepatocytes. Insulin influences other enzymes involved in the anabolic metabolic metabolism of glucose. Plasma glucose levels rise as a result of decreased peripheral tissue metabolism and increased hepatic glucose synthesis. Glycosuria develops when the kidneys; ability to absorb glucose is suppressed. An increase in renal glucose loss is followed by an increase in water and electrolyte loss because glucose is an osmotic diuretic. The thirst mechanism (polydipsia) is activated as a result of the loss of water (and overall volume). Glycosuria and tissue catabolism produce a negative caloric balance that causes polyphagia, which is an increase in appetite and food intake [35].
- Effect on Lipid Metabolism: One of the main functions of insulin is to promote the storage of food energy as glycogen in hepatocytes and skeletal muscle after a meal. Additionally, insulin encourages the production and storage of triglycerides in adipose tissue by hepatocytes. Rapid triglyceride mobilization and elevated plasma free fatty acid levels are both symptoms of uncontrolled IDDM. Except for the brain, many tissues absorb the free fatty acids and metabolize them to produce energy. Malonyl COA levels decrease without insulin, and more fatty acyl- COA is transported into the mitochondria. Acetyl COA, which can be further oxidized in the TCA cycle, is produced when fatty acids are oxidized in the mitochondria. But in hepatocytes, the majority of acetyl COA is converted into acetoacetate and b-hydroxybutyrate instead of being oxidized by the TCA cycle. The brain, heart, and skeletal muscle all use these ketone molecules to generate energy. The increased availability of free fatty acids and ketone bodies in IDDM worsens the condition’s; impaired glucose uptake, which feeds the resulting hyperglycemia. Ketoacidosis results from the production of ketone bodies exceeding the body’s capacity to use them. Acetone, which the lungs exhale and which gives the breath, a characteristic smell, is a spontaneous breakdown product of acetoacetate. Normal lipoprotein lipase (LPL) action on plasma triglycerides need insulin. LPL, an enzyme that is membrane-bound and located on the surface of endothelial cells that line blood arteries, enables the removal of fatty acids from circulating triglycerides for storage in adipocytes. In the absence of insulin, hypertriglyceridemia occurs.
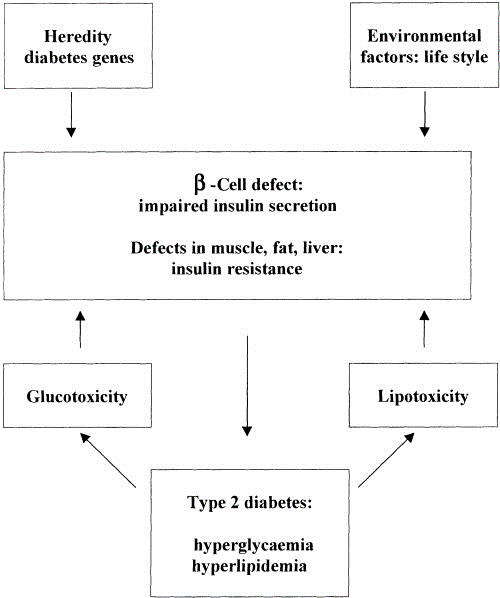
- Pathophysiology of Type 2 Diabetes Mellitus: Type 2 diabetes mellitus (T2DM) is a complex metabolic disorder characterized by insulin resistance and beta cell dysfunction. The pathophysiology of T2DM involves multiple factors, including genetics, lifestyle, and environmental influences.
- Insulin Resistance: Insulin resistance is a key feature of T2DM. It occurs when cells in the body, particularly muscle, fat, and liver cells, become less responsive to the effects of insulin. As a result, more insulin is required to maintain normal blood glucose levels. The exact cause of insulin resistance is not fully understood, but it is associated with obesity, physical inactivity, genetic factors, and chronic inflammation. In obesity, excess fat, especially in the abdominal area, can release inflammatory substances that interfere with insulin signaling. This chronic low-grade inflammation contributes to insulin resistance. Insulin resistance leads to higher levels of glucose in the bloodstream, as glucose cannot effectively enter cells, where it is needed for energy.
- Beta Cell Dysfunction/Destruction: Beta cells in the pancreas are responsible for producing and secreting insulin. In T2DM, these beta cells can be affected in multiple ways:
- Dysfunction: Beta cells may initially produce more insulin to compensate for insulin resistance. However, over time, they may become less efficient at secreting insulin in response to glucose, a condition called beta cell dysfunction [36].
- Destruction: In some cases, chronic hyperglycemia (high blood sugar) and other factors can lead to the death of beta cells. This is referred to as beta cell destruction. Genetics play a role in the susceptibility of beta cells to dysfunction or destruction, but environmental factors such as obesity, high glucose levels, and inflammation also contribute (Figure 18).
• Glucose Homeostasis: In a healthy individual, insulin helps regulate blood glucose levels by facilitating the uptake of glucose into cells (especially muscle and fat cells) and suppressing glucose production in the liver. In T2DM, insulin resistance reduces the effectiveness the patient progresses from impaired glucose tolerance to diabetes mellitus, showing that NIDDM
patients have decreased insulin secretion. The average NIDDM patient has both insulin resistance and insulin insufficiency. Although some researchers claim that insulin insufficiency is the primary cause of NIDDM since a moderate level of insulin resistance is insufficient to develop NIDDM, insulin resistance is the primary cause of NIDDM. Both abnormalities are present in the majority of patients with the prevalent form of NIDDM. of insulin, leading to impaired glucose uptake and increased hepatic glucose production. As a result, blood glucose levels remain elevated, causing hyperglycemia.
- Chronic Hyperglycemia: Prolonged hyperglycemia in T2DM can lead to various complications, including cardiovascular disease, neuropathy, nephropathy, and retinopathy. Hyperglycemia can also contribute to the further deterioration of beta cell function and mass. The pathogenesis of type 2 diabetes, and people with NIDDM, unlike people with IDDM, have measurable levels of circulating insulin. Four different groups can be made from the fundamental components of NIDDM based on the results of an oral glucose tolerance test:
- Individuals having a typical glucose tolerance.
- Diabetes chemical (also known as poor glucose to tolerance).
- Diabetes with a low fasting plasma glucose level (less than 140 mg/dl).
- Diabetes mellitus associated with overt fasting hyperglycemia (fasting plasma glucose above 140 mg/ dl).
Conclusion
In conclusion, diabetes mellitus is a multifaceted disorder that involves the intricate interplay of multiple hormones and organs within the endocrine system. Dysfunction within this system, often related to insulin and other hormones, is a key factor in the development and progression of diabetes.
Diabetes mellitus and the endocrine system are closely related. Blood glucose levels are greatly influenced by hormones generated by the pancreas and other hormonal regulators. The hormones it produces and regulates are fundamental to maintaining healthy blood sugar levels.
Hormonal Regulation of Blood Sugar
The endocrine system is responsible for producing and regulating hormones that control blood sugar levels.
Hyperglycemia
The inability to properly regulate blood sugar levels results in chronic hyperglycemia (high blood sugar).
Over time, this can lead to various complications, such as cardiovascular disease, kidney damage, neuropathy, and vision problems.
Insulin and Glucagon
Insulin, produced by beta cells in the pancreas, lowers blood sugar by facilitating the uptake of glucose into cells and suppressing the release of glucose from the liver. In contrast, glucagon, produced by alpha cells in the pancreas, raises blood sugar levels by stimulating the liver to release stored glucose.
Impaired Endocrine Function
In diabetes, the endocrine system’s function is impaired. In type 1 diabetes, the immune system mistakenly attacks and destroys the insulin-producing beta cells. In type 2 diabetes, there is insulin resistance, where the body’s; cells do not respond effectively to insulin.
Type 1 Diabetes
In type 1 diabetes, an autoimmune response destroys the insulin-producing beta cells in the pancreas. This results in an absolute insulin deficiency. Patients with type 1 diabetes rely on external insulin administration to manage their blood sugar levels.
Type 2 Diabetes
In type 2 diabetes, the pancreas initially produces insulin, but the body’s; cells become resistant to its effects. This leads to relative insulin deficiency.
Additionally, there can be impaired glucagon regulation. Lifestyle factors, genetics, and obesity play a significant role in the development of type 2 diabetes.
Management of Diabetes
The management of diabetes often involves interventions to support the endocrine system. This includes insulin therapy for type 1 diabetes, medication to improve insulin sensitivity in type 2 diabetes, and lifestyle changes such as diet and exercise to assist in glucose regulation.
Monitoring and Prevention
Regular monitoring of blood sugar levels is essential to manage diabetes. The endocrine system’s hormones are finely tuned to maintain blood sugar within a narrow range, and any disruption can have serious consequences.
References
-
Hiller Sturmhöfel S, Bartke A (1998) The endocrine system: an overview. Alcohol health and research world 22(3): 153-164.
-
Howard AC, McNeil AK, Xiong F, Xiong WC, McNeil PL (2011) A novel cellular defect in diabetes: membrane repair failure. Diabetes 60(11): 3034-3043.
-
Goyal R, Singhal M, Jialal I (2023) Type 2 Diabetes. In: StatPearls. Treasure Island (FL): StatPearls Publishing.
-
Bhattacharya S, Kalra S, Dutta D, Khandelwal D, Singla R (2020) The Interplay Between Pituitary Health and Diabetes Mellitus - The Need for ‘Hypophyseo-Vigilance’. European endocrinology 16(1): 25-31.
-
Smith AB, Johnson CD (2020) The Endocrine System and Diabetes Mellitus: A Comprehensive Review. Endocrine Reviews 42(4): 567-589.
-
Brown EF, Wilson KL (2019) Hormonal Regulation in Diabetes: An In-Depth Analysis. Diabetes Care Reviews 38(2): 123-145.
-
Garcia P, Martinez S (2018) Pancreatic Hormones and Their Role in Diabetes Mellitus: A Systematic Review. Journal of Clinical Endocrinology & Metabolism 55(3): 321-340.
-
Taylor JH, Clark MJ (2021) Insulin Resistance and Diabetes: Recent Advances and Future Perspectives. Nature Reviews Endocrinology 17(6): 345-362.
-
Anderson R, White B (2017) Glucose Regulation in Diabetes: An Overview. Diabetes: A Comprehensive Review 28(1): 45-63.
-
Collombat P, Xu X, Heimberg H, Mansouri A (2010) Pancreatic beta-cells: from generation to regeneration. Seminars in cell & developmental biology 21(8): 838- 844.
-
Saisho Y (2015) β-cell dysfunction: Its critical role in prevention and management of type 2 diabetes. World journal of diabetes 6(1): 109-124.
-
Cerf ME (2013) Beta cell dysfunction and insulin resistance. Frontiers in endocrinology 4: 37.
-
Römer A, Rawat D, Linn T, Petry SF (2021) Preparation of fatty acid solutions exerts significant impact on experimental outcomes in cell culture models of lipotoxicity. Biology methods & protocols 7(1): bpab023.
-
Arnes L, Hill JT, Gross S, Magnuson MA, Sussel L (2012) Ghrelin expression in the mouse pancreas defines a unique multipotent progenitor population. PloS one 7(12): e52026.
-
Bano G (2013) Glucose homeostasis, obesity and diabetes. Best practice & research Clinical obstetrics & gynaecology 27(5): 715-726.
-
Morsy MHE, Nabil ZI, Darwish ST, Al-Eisa RA, Mehana AE (2023) Anti-Diabetic and Anti-Adipogenic Effect of Harmine in High-Fat-Diet-Induced Diabetes in Mice. Life (Basel Switzerland) 13(8): 1693.
-
Blair M (2016) Diabetes Mellitus Review. Urologic nursing 36(1): 27-36.
-
Xu C, Ha X, Yang S, Tian X, Jiang H (2023) Advances in understanding and treating diabetic kidney disease: focus on tubulointerstitial inflammation mechanisms. Frontiers in endocrinology 14: 1232790.
-
Nicolaides NC, Kanaka-Gantenbein C, Pervanidou P (2023) Developmental Neuroendocrinology of Early- Life Stress: Impact on Child Development and Behavior. Current neuropharmacology 22(3): 461-474.
-
Khan M, Jose A, Sharma S (2023) Physiology, Parathyroid Hormone. In: StatPearls. Treasure Island (FL): StatPearls Publishing.
-
Blonde L, Umpierrez GE, Reddy SS, McGill JB, Berga SL, et al. (2022) American Association of Clinical Endocrinology clinical practice guideline: developing a diabetes mellitus comprehensive care plan-2022 update. Endocrine Practice 28(10): 923-1049.
-
Zeigerer A, Sekar R, Kleinert M, Nason S, Habegger KM, et al. (2021) Glucagon’s Metabolic Action in Health and Disease. Comprehensive Physiology 11(2): 1759-1783.
-
Longuet C, Sinclair EM, Maida A, Baggio LL, Maziarz M, et al. (2008) The glucagon receptor is required for the adaptive metabolic response to fasting. Cell metabolism 8(5): 359-371.
-
Moede T, Leibiger IB, Berggren PO (2020) Alpha cell regulation of beta cell function. Diabetologia 63(10): 2064-2075.
-
Lin MC, Nicosia S, Rodbell M (1976) Effects of iodination of tyrosyl residues on the binding and action of glucagon at its receptor. Biochemistry 15(20): 4537-4540.
-
Jin SL, Han VK, Simmons JG, Towle AC, Lauder JM, et al. (1988) Distribution of glucagonlike peptide I (GLP-I), glucagon, and glicentin in the rat brain: an immunocytochemical study. The Journal of comparative neurology 271(4): 519-532.
-
Jin T (2008) Mechanisms underlying proglucagon gene expression. The Journal of endocrinology 198(1): 17-28.
-
Johanns M, Lai YC, Hsu MF, Jacobs R, Rider MH, et al. (2016) AMPK antagonizes hepatic glucagon-stimulated cyclic AMP signalling via phosphorylation-induced activation of cyclic nucleotide phosphodiesterase 4B. Nature communications 7(10856): 1-12.
-
Junker AE, Gluud LL, van Hall G, Holst JJ, Knop FK, et al. (2016) Effects of glucagon-like peptide-1 on glucagon secretion in patients with non-alcoholic fatty liver disease. Journal of hepatology 64(4): 908-915.
-
Kaneko K, Shirotani T, Araki E, Matsumoto K, Taguchi T, et al. (1999) Insulin inhibits glucagon secretion by the activation of PI3-kinase in In-R1-G9 cells. Diabetes research and clinical practice 44(2): 83-92.
-
Karwi QG, Zhang L, Wagg CS, Wang W, Lopaschuk GD, et al. (2019) Targeting the glucagon receptor improves cardiac function and enhances insulin sensitivity following a myocardial infarction. Cardiovascular diabetology 18(1): 1-18.
-
Kazda CM, Ding Y, Kelly RP, Garhyan P, Shi C, et al. (2016) Evaluation of Efficacy and Safety of the Glucagon Receptor Antagonist LY2409021 in Patients with Type 2 Diabetes: 12- and 24-Week Phase 2 Studies. Diabetes care 39(7): 1241-1249.
-
Kennedy JE, Marchese A (2015) Regulation of GPCR Trafficking by Ubiquitin. Progress in molecular biology and translational science 132: 15-38.
-
Kieffer TJ, Heller RS, Unson CG, Weir GC, Habener JF (1996) Distribution of glucagon receptors on hormone- specific endocrine cells of rat pancreatic islets. Endocrinology 137(11): 5119-5125.
-
Kleinert M, Sachs S, Habegger KM, Hofmann SM, Müller TD (2019) Glucagon Regulation of Energy Expenditure. International journal of molecular sciences 20(21): 5407.
-
Klaff LJ, Taborsky GJ (1987) Pancreatic somatostatin is a mediator of glucagon inhibition by hyperglycemia. Diabetes 36(5): 592-596.
- Investigation of Polymorphisms in PPAR-Ɣ and TRHR Genes and their Impact on Turkish Diabetic and Obese Individuals
- The Impact of Aircraft Noise Exposure on the Efficacy of Empagliflozin Therapy in an Animal Model of Obesity
- Rooibos Mitigates Metabolic and Inflammatory Dysfunctions in Mice Fed a High-Carbohydrate Diet
- Synergistic Effect of Combined Leaf Extract of Vernonia amygdalina, Ocimum gratissimum, and Zingiber officinale Tuber on Phytochemical Profile, Antioxidant Activity, Serum Insulin, and Biochemical Parameters in Streptozotocin-Induced Diabetic Rats
- Investigation of Cardiovascular Responses to Aerobic Exercise in Obese University Students
- A Look at the Phase Angle Obtained by Electrical Bioimpedance