S-Nitrosoglutathione Stabilizes Mature, Cell Surface CFTR through Aactivator of the Hsp90 ATPase
Introduction: Cystic fibrosis (CF) is a multisystem disease associated with mutations in the gene that encodes the CF transmembrane conductance regulatory (CFTR) protein. S-nitrosothiols (SNOs) are endogenous cell signaling molecules with relevance to human lung disease, including cystic fibrosis (CF). S-nitrosoglutathione (GSNO), one class of SNOs levels are significantly lower in the CF airway than the healthy airway. Here, we address whether GSNO interact with activator of the Hsp90 ATPase (Aha1), which targets improperly folded CFTR for degradation. Materials and methods: We used human bronchial airway epithelial (CFBE410-) cells expressing wild type and mutant F508del CFTR protein. Immunoblot analysis was performed in the presence or absence of GSNO by using monoclonal antibodies for Aha1 and CFTR. We knocked down endogenous Aha1 with Aha1 siRNA duplexes. Next, we tested Aha1 S-nitrosylation by using a biotin switch assay and measurement of ATPase activity by using the EnzChek phosphate detection kit. Finally, we used confocal laser scanning microscopy for co-localization of Aha1 and CFTR. Results: Our study showed that GSNO significantly decreased Aha1 expression. Following Aha1 knockdown, CFTR maturation levels were elevated at the cell surface in F508del CFTR. In the presence of GSNO. F508del CFTR maturation was further increased. We found that CFTR-associated Aha1 is S-nitrosylated and inhibits its ATPase activity by GSNO. Finally, we showed cellular co-localization of Aha1 with CFTR by confocal microscopy. Conclusion: Our observations offer a novel approach in identifying the key mechanism by which SNOs increase F508del CFTR maturation and cell membrane stabilization.
Introduction
Cystic fibrosis (CF) is an inherited disorder that is especially common among those with European heritage. The
disease affects multiple organ systems by thickening epithelia mucus into a thick copious substance that accelerates host infection [1]. The genetic component of CF is varied and has been associated with roughly 2,000 mutations in the CFTR gene. CFTR is an epithelial chloride channel regulated by cGMP and a subtype of the ABC- transporter superfamily that is expressed in the luminal membrane of epithelial cells in the airways, pancreas and other organs [2]. In F508del- CFTR, the most common genetic mutation associated with CF, CFTR folds incorrectly because a phenylalanine is deleted from the CFTR amino acid sequence. Misfolded CFTR cannot fully mature and is degraded by molecular chaperones before it reaches the cell surface [3, 4], causing the loss of epithelial Cl- conductance and organ damage. F508del CFTR present at the cell membrane is partially functional, so there is interest in CF treatments that would allow misfolded CFTR to reach the cell surface [4, 5]. S-nitrosothiols (SNOs) are endogenous signaling molecules with many useful effects on the human airways [6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16]. Multiple proteins relevant in pulmonary disease, including CF, undergo S-nitrosylation reactions, which are metabolically regulated post-translational modifications that avert the degradation of specific proteins [8, 9, 10, 11]. We have been interested in examining whether SNOs prevent CFTR degradation and increase its maturation. Different SNOs increase the expression and function of both wild type and mutant F508del CFTR [17]. SNOs can increase CFTR maturation by S-nitrosylating specific cysteine residues in the co-chaperones or molecular chaperones involved in the trafficking and biogenesis of CFTR, such as heat shock protein (Hsp) 70, Hsp90 organizing protein (Hop), heat shock cognate 70 (Hsc70), cysteine string protein (Csp) and C-terminus Hsc70 interacting protein (CHIP) [18, 19, 20, 21, 22, 23, 24, 25, 26, 27]. Molecular chaperones are proteins that help other proteins fold, but they do not become part of the final product [27, 28, 29, 30, 31, 32]. SNOs increase CFTR maturation by S-nitrosylating cysteine residues on heat shock protein (Hsp) 70, heat shock cognate 70 (Hsc70), Hsp90 organizing protein (Hop) and cysteine string protein (Csp) [5, 19, 20].
Aha1 (38 kDa) is an essential ubiquitous Hsp90 co- chaperone and an ATPase regulator. An interaction between Aha1 and Hsp90 is crucial for allowing CFTR to fold properly [28, 29, 30, 31, 32, 33] and is believed to play a critical role in directing folding-deficient F508del CFTR for degradation [4, 27, 28, 29, 30, 31, 32, 33]. Inhibiting the formation of the Hsp90-Aha1 chaperone complex formation would prevent proteolysis of the Hsp90-F508del CFTR complex and allow it to function as a chloride channel [33]. In this work we demonstrate that S-nitrosoglutathione (GSNO) reduced both Aha1 expression and function. Mimicking the effects of SNOs, Aha1 knock- down with siRNA leads to greater F508del CFTR expression at the cell surface. Targeting this SNO-Aha1 interaction has potential as an innovative CF corrector.
Materials and Methods
Chemicals and Reagents
Pepstatin A and electrophoresis reagents were obtained from Boehringer Ingelheim (Indianapolis, IN) and Bio- Rad (Hercules, CA), respectively. Leupeptin and aprotinin were acquired from Roche Diagnostics (Mannheim, Germany). Unless otherwise, all other chemicals were purchased from Sigma Chemical Company (St. Louis, MO). S-nitrosoglutathione (GSNO) was prepared according to previously published protocol [11, 19].
Cell Culture
We cultured CF bronchial epithelial (CFEB41o-) cell lines provided by Dr. Eric Sorscher (University of Alabama Birmingham, AL) expressing wild type & homozygous mutant F508del CFTR. Cells were grown in a monolayer at 37°C in humidified 5% CO2 air using Dulbecco’s modified Eagle medium (DMEM) as described before [17, 18, 19, 20, 21].
Western Blotting and Immunoprecipitation
Western blotting was completed following previously published protocol [17, 18, 19, 20, 21]. Whole cell extracts were prepared in 1% NP-40 lysis buffer (50 mM Tris-HCl, pH 8.0, 1% NP-40, 150 mM NaCl, 2 µM leupeptin, 1 µM aprotinin, and 1 µM pepstatin, 1mM DTT, 1 µM PMFS, and 2 µM Na3VO4). After centrifugation at 10,000 RPM to recover insoluble materials, cellular materials were sheared by passing through a 25-guage needle. After extraction, we performed a Lowry assay (BCA Protein Assay Kit, Pierce Protein Research Products, Thermo Fisher Scientific, Waltham, MA). 100 µg of protein were fractionated on a 6% SDS polyacrylamide gel under reducing conditions and then shifted to nitrocellulose membranes (Bio-Rad, Hercules, CA). After blots were blocked in Tris buffered saline-Tween 20 containing 5% nonfat dried milk, they were probed with a 1:1000 dilution of anti-CFTR mAb 596 antibody (University of North Carolina, Chapel Hill, NC) or a 1:1000 dilution of Aha1 antibody, anti-Aha1 (Thermo Fisher Scientific) and a 1:3000 dilution of HRP-conjugated secondary antibody (Santa Cruz Biotechnologies). Once the blots were washed, they were visualized using enhanced chemiluminescence (ECL, Super Signal West Pico, Chemiluminescent Substrate, Thermo Fisher Scientific). Blots were stripped and probed with a 1:1000 dilution of anti-β-actin antibody, rabbit monoclonal IgM (Cell Signaling Technology, Danvers, MA) as a loading control. Using Quantity One software (Bio-Rad), we determined relative protein quantitation of each band using densitometry. We performed immunoprecipitation (IP) as previously outlined [19]. Confluent CFBE41o- grown in monolayer expressing wild-type and mutant F508del CFTR
were used. Whole cell extracts were prepared as outlined above for the Western blot analysis. Ten µl of primary Aha1 antibody (anti-Aha1, Thermo Fisher Scientific) or CFTR antibody (anti-CFTR mAb 596 antibody, University of North Carolina Chapel Hill) were added to each fraction, and they were incubated overnight at 4°C with gentle shaking. After supernatant antibodies mixtures were treated with 70 µl of Protein A (Boehringer Mannheim, Indianapolis, IN), they were incubated for another 4 h. After the samples were centrifuged for one min, they were washed twice with RIPA buffer to remove bound proteins from beads. Proteins were eluted from the beads using 100 µl of sample buffer, incubated and mixed for one h. After IP, 50 µg of protein were fractionated on a 6% SDS polyacrylamide gel under reducing conditions as outlined above.
Cell Surface Protein Biotinylation
Cell surface biotinylation was carried out as previously outlined [18, 19]. CFBE41o- cells expressing mutant F508del CFTR were exposed to the indicated GSNO concentrations for 4h. After the cells were washed three times with ice-cold PBS containing 0.1 mM CaCl2 and 1 mM MgCl2 (PBSCM), they were treated in the dark with PBSCM buffer containing 10 mM sodium periodate at 20°C for 30 min. The cells were washed three more times with PBSCM and biotinylated by treating them with sodium acetate buffer (100 mM sodium acetate buffer, pH 5.5; 0.1 mM CaCl2 and 1 mM MgCl2) containing 2 mM biotin-LC hydrazide (Pierce, Rockford, IL) for 30 min at 20°C in the dark. The cells were washed three times with sodium acetate buffer and solubilized using lysis buffer with protease inhibitors and Triton X-100 and. CFTR was immunoprecipitated as previously described [19] and subjected to SDS-PAGE on 6% gels under reducing conditions. Streptavidin-conjugated horseradish peroxidase was used to detect biotinylated CFTR.
Knock-down of endogenous Aha1 by siRNA
HPLC analysis (Santa Cruz Biotechnologies, Dallas, TX) revealed the Aha1-siRNA duplexes exhibited >90% purity. Scrambled siRNA served as the control group. F508del CFTR CFBE41o- cells were transfected with 50 nM of Aha1 siRNA or scrambled. 48h later, they were washed three times with PBS and then lysed. A 100 µg sample of protein was fractionated on a 6% SDS-PAGE. Blots were probed at a 1:500 dilution of monoclonal anti-Aha1 antibody (Thermo Fisher Scientific) and HRP-conjugated secondary antibody to confirm positive knockdown.
S-Nitrosylation of Co-chaperone Aha1 and Measurement of ATPase Activity
S-Nitrosylation of Aha1 was carried out as defined before [18, 19]. CFBE41o- cells were exposed to 5 and 10 µM GSNO or buffer for 4 h in humidified 5% CO2 air at 37°C, and proteins were extracted without reducing agents. S-nitosylated proteins were isolated from 100 µg of each extract by the biotin switch method [19]. Whole cell lysates were precipitated using acetone and re-suspended in 100 µl HEN Buffer (250 mM HEPES pH 7.7, 1 mM EDTA, 0.1 mM neocuproine). Samples were mixed with 4 volumes of blocking solution and incubated at 50°C for 20 min with shaking. Protein was precipitated by acetone, re- suspended in HEN buffer containing 1 mM Biotin-HPDP (Pierce, Rockford, IL) and 1 mM ascorbate and incubated for 1 h at room temperature. Biotin HPDP was removed by precipitation with acetone and the pellet was re-suspended in 100 µl HEN buffer and neutralized using neutralization buffer (20 mM HEPES pH 7.7, 10 mM NaCl, 1 mM EDTA, 0.5% Triton X-100.). Biotinylated proteins were isolated by incubation with streptavidin agarose for 1 h at room temperature. Resin was washed five times with neutralization buffer containing 600 mM NaCl and biotinylated protein was eluted with SDS-PAGE loading buffer. Protein ATPase activity was measured using the EnzChek phosphate detection kit (Molecular Probes, Eugene, Oregon). Wild type and F508del CFBE41o- cells were treated with 5 and 10 µM GSNO for 4h and then immunoprecipited for SNO-Aha1. 120 µl cuvettes were used for the assay. A Baeckman Coulter DU 530 spectrophotometer (Indianapolis, IN) was used to determine phosphate production as a measure of baseline Pi of solution and Pi produced by ATPase by measuring absorbance at 360 nm. Assays were performed in 40 mM HEPES, pH 7.5, 150 mM KCl, 5 mM MgCl2, and 2 mM ATP at 37°C.
Immunofluorescence and Confocal Microscopy
Immunofluorescence and confocal laser scanning microscopy were carried out as described before [34, 35]. Once CFBE41o- cells were harvested to confluence on glass coverslips, they were rinsed three times with 5 ml of PBS, fixed with 3.7% paraformaldehyde for 5 min and then rinsed three times with PBS, permeabilized with 0.1% Triton X-100 in PBS (v/v) (10 min), rinsed two more times with PBS, then incubated with blocking solution of 5% normal goat serum (NGS) and 0.01% Triton X-100 in 1X PBS (PBS-T-NGS) for 45 min at room temperature and incubated overnight at 4ᵒC with primary anti-Aha1 antibody (1:100, anti-Aha1,Thermo Fisher Scientific) or with primary anti-CFTR antibody (1:150 dilution, mouse monoclonal mAb 596, University of North Carolina Chapel Hill). The cells were then rinsed with blocking solution twice for 15 min and incubated at room temperature for 45 min with a secondary antibody to CFTR (1:500 dilution, Alexa Fluor 568, Molecular Probes, Invitrogen, Carlsbad, CA) or Aha1 (1:500 dilution, Alexa Flour 488, Molecular Probes, Invitrogen), made in blocking solution. The cells were washed twice for 15 min in blocking solution with 0.1% NGS, stained with DAPI for nuclear staining, and washed in PBS. As a negative control, an isotype control for mouse and rabbit (BD Pharmingen, Mississauga, ON Canada) were respectively used at the same concentration as Aha1 and CFTR primary antibodies, followed by labeling with the individual secondary antibodies. The cells were mounted and imaged using 63x NA1.4 oil immersion lens on Leica TCS SP5 for laser scanning confocal microscopy. The white light lasers of 488 nm (20%) and 568nm (65%) were used for excitation of Aha1-Alexa488 (Emission 500-550 nm) and CFTR-Alexa 568 (Emission 580-640 nm) correspondingly. Alike imaging parameters were used for isotype controls.
Statistics
A two-tailed unpaired t-test was conducted and results are stated as means ± SD. p < 0.05 was considered significant.
Results
GSNO decreases the endogenous Aha1 levels in CFBE41o- cells
Both mutant F508del CFTR and wild type CFBE41o- cells expressed Aha1 (Figure 1A). We exposed CFBE41o- cells to 10 µM of GSNO, which was previously shown to be effective [17, 18, 19, 20, 21, 22, 23, 24, 25, 26]. After 4h, we found decreased Aha1 expression in these cells (4.0-fold, and 3.6-fold, p<0.002; n=4; Figures 1A, B).
Aha1 Knockdown Promotes F508del CFTR Expression
CFBE41o- cells expressing F508del CFTR were A
GSNO (10 µM) - + - +
$$
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - F508del Wt
4
| * | * | ||||||||||
B
3 Fold Change
2
1 Figure 1: Aha1 is expressed in CFBE41o- cells expressing mutant F508del and wild type CFTR by Western blot using rabbit polyclonal anti-Aha1 antibody and 100 µg protein load (A). In the presence or absence of 10 µM GSNO for 4 h. We show 10 µM GSNO decreased the Aha1 expression levels in both mutant and wild type cells. Membranes were stripped and reprobed with β-actin. The optical densities of the bands were quantified by densitometry (B). Data are the mean ±SD, n=3, *p<0.01.
transfected with 50 nM of siRNA Aha1 duplexes specific for Aha1. We found that cell surface expression of F508del CFTR increased 48h after transfection (2.6-fold, p<0.002, n=3). siRNA transfected CFBE41o- cells incubated with GSNO (10 µM, 4h) further increased F508del maturation compared to non-treated cells (3.5-fold, p<0.002, n=3). When a scrambled siRNA control was applied, CFTR expression was not affected (Figures 2A, B).
CFTR-associated Aha1 is S-nitrosylated and Inhibits its ATPase Activity
We checked whether the F508del CFTR-associated Aha1 is S-nitrosylated. CFBE41o- cells were exposed to 5 and 10 µM GSNO for 4 hours. To co-immunoprecipitate Aha1 and CFTR, isolated whole cell extracts were incubated with antisera. An established biotin switch assay was used to measure SNO-modified Aha1 proteins as previously outlined [19]. We found that Aha1 is S-nitrosylated in a concentration- dependent manner. Densitometry was used to measure the optical densities of the bands (Figure 3B, n=3, p<0.01). Wild type and mutant F508del CFBE41o- cells were treated with 5 and10 μM GSNO for 4 h, IP for SNO-Aha1, Protein ATPase activity was then quantified in both conditions. In both wild type and F508del CFBE41o- cells, 10 µM GSNO significantly decreased ATPase activity (1.2-fold and 1.77- fold respectively, p<0.01, n=3, Figure 3C)
Cellular Co-localization of Aha1 and CFTR
Immunofluorescence microscopy revealed Aha1 partially co-localizes with CFTR (Figure 4A). When isotype controls were examined, no signal was observed (Figure 4B), supporting the notion that the co-chaperone Aha1 interacts with CFTR in CFBE41o- cells.
Aha1
β-actin
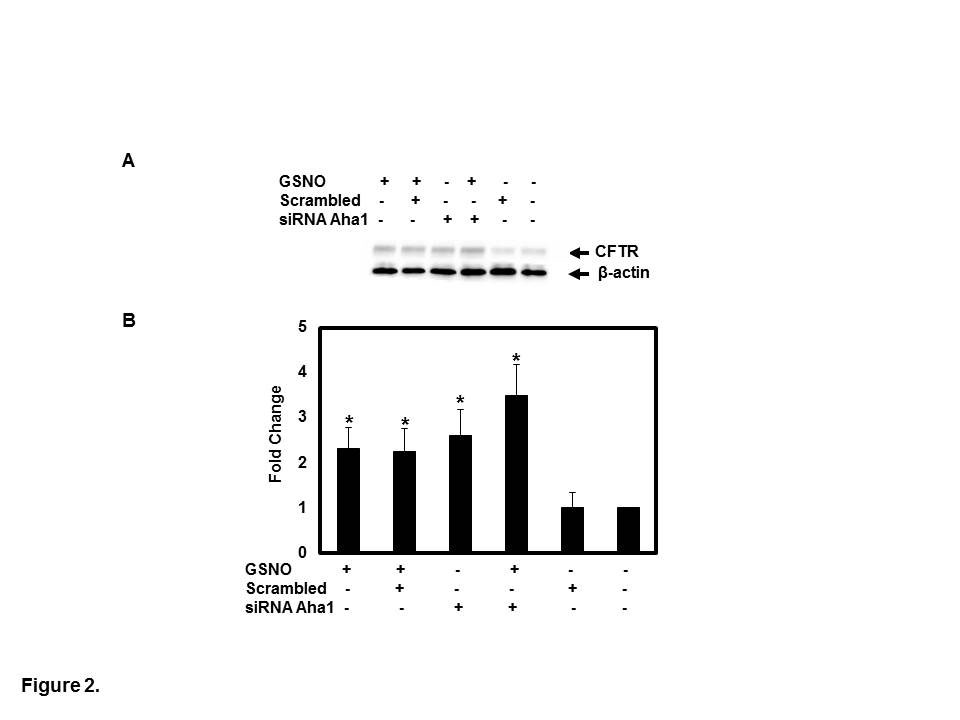
Figure 2: Knock down of endogenous Aha1 was accomplished by transfection of CFBE41o- cells with 50 nM of Aha1 siRNA duplexes specific for Aha1compared to scrambled. After 48 h post-transfection, cells were treated for 4 h in the presence or absence of GSNO. Cell surface biotinylation was performed, as previously described [19]. CFTR was IP with monoclonal antibody (anti-CFTR mAb596) and subjected to SDS-PAGE on 6% gels; biotylated CFTR was detected with streptavidin- conjugated HRP. The membrane was stripped and reprobed with β-actin (A). The optical densities of the bands were quantified by densitometry (B). Data are the mean ±SD, n=3, *p<0.01.
![Figure 2: Knock down of endogenous Aha1 was accomplished by transfection of CFBE41o- cells with 50 nM of Aha1 siRNA duplexes specific for Aha1compared to scrambled. After 48 h post-transfection, cells were treated for 4 h in the presence or absence of GSNO. Cell surface biotinylation was performed, as previously described [19]. CFTR was IP with monoclonal antibody (anti-CFTR mAb596) and subjected to SDS-PAGE on 6% gels; biotylated CFTR was detected with streptavidin- conjugated HRP. The membrane was stripped and reprobed with β-actin (A). The optical densities of the bands were quantified by densitometry (B). Data are the mean ±SD, n=3, *p<0.01.](/fulltextimages/5880/fig_2.png)
Figure 3: CFBE41o- cells were exposed to 5 and 10 µM GSNO for 4 hr. SNO proteins in whole cell lysates underwent biotin switch assay, followed by streptavidin purification and underwent immunoblot with anti-Aha1 antibody (A), as described previously [27]. The optical densities of the bands were quantified by densitometry (B). GSNO modification of Aha1 ATPase activity (C), was measured using the EnzChek phosphate detection kit. Wild type and F508del CFBE41o- were treated with 5 and 10 μM GSNO for 4 h and IP SNO-Aha1, then protein ATPase activity was measured in both conditions (C). Data are the mean ±SD, n=3, *p<0.01.
CFTR Aha1 Merge
![Figure 3: CFBE41o- cells were exposed to 5 and 10 µM GSNO for 4 hr. SNO proteins in whole cell lysates underwent biotin switch assay, followed by streptavidin purification and underwent immunoblot with anti-Aha1 antibody (A), as described previously [27]. The optical densities of the bands were quantified by densitometry (B). GSNO modification of Aha1 ATPase activity (C), was measured using the EnzChek phosphate detection kit. Wild type and F508del CFBE41o- were treated with 5 and 10 μM GSNO for 4 h and IP SNO-Aha1, then protein ATPase activity was measured in both conditions (C). Data are the mean ±SD, n=3, *p<0.01.](/fulltextimages/5880/fig_3.jpeg)
a
Mouse Isotype control Rabbit Isotype control
b
Figure 4: Confocal microscopy was performed on CFBE41o- cells. First cells were fixed with 3.7% paraformaldehyde for 5 minutes, permeabilized by 0.1% Triton X-100 and incubated with primary CFTR (anti-CFTR mAb 596) and anti-Aha1 antibody at 4oC overnight. Cells were incubated with secondary antibodies for 45 minutes at RT, then mounted with coverslips and visualized with the Leica laser scanning confocal microscopy. Representative images are shown from triplicate experiments revealing co-localization of CFTR (green) and Aha1 (red) in merged images (orange) (a). The mouse and rabbit isotypes were used as a negative control (b).
Discussion
Cystic fibrosis can manifest as frequent respiratory infections, chronic lung disease and death. In patients with F508del, the CFTR protein is not on the plasma membrane because it is degraded before fully maturing, but it can still operate as a less effective chloride channel if it can be driven to the plasma membrane [2, 3, 4]. Finding improvements to therapies that can bring F508del CFTR to the plasma membrane is crucial.
With low concentrations of SNOs, mutant F508del CFTR maturation and cell membrane expression increases [19, 21]. S-nitrosylating agents, including GSNO, influence CFTR expression by activating the specificity protein, Sp1/ Sp3 transcription factors [18] and participating in intricate interactions between various co-chaperones that affect the folding and trafficking of F508del CFTR [19]. S-nitrosylating the cysteine residues of specific chaperones that regulate CFTR biogenesis and cell surface trafficking increases CFTR expression [19, 20, 27, 28, 29, 30, 31, 32].
SNO levels in the airways are lowered CF patients [9]. Thus impending degradation of GSNO itself is an ineffective treatment for CF airway disease. Inhibition of
50µm
50µm S-nitrosoglutathione reductase (GSNO-Reductase) failed to improve lung function in CF patients, but it lowered sweat chloride levels and increased weight gain likely because GSNO synthesis is decreased in [22], Exogenous GSNO has been used safely in human trials, and it has numerous benefits independent of CFTR [18, 19, 20, 21, 22, 24, 25, 26]. Thus direct replacement of GSNO or an equivalent will likely be more effective than inhibiting catabolism [34].
Transnitrosation is required in the signaling pathways of many different cell and organ systems. We have examined whether compounds that can bypass the bioactivation required for GSNO can act as intracellular NO- donors [18, 19, 20]. Dose-response studies conducted in cell cultures treated with 5-10 μM of GSNO is ideal for correcting CFTR in CF cell monolayers [19, 21]. However, transnitrosation augments CFTR maturation so inhalational SNO-like agents could potentially serve as effective F508del correctors. In full-thickness cells, GSNO analogues that are cell permeable can increase functional F508del CFTR expression [20]. Thus the effects of S-nitrosylation could allow F508del CFTR to mature in complex epithelial systems if the cell membrane permeable ester is used [19]. Some potentially relevant therapies that should considered include inhaled GNOD, SNOAC, and ethyl nitrate.
Previous studies demonstrated that CFTR trafficking is regulated by Aha1 [29, 30, 31, 32, 33]. We demonstrated that Aha1 is expressed in both CFBE41o- cells expressing both mutant F508del CFTR and wild type CFTR. Note that expression levels were lower in CFBE41o- cell lines (both F508del/ wild-type) when compared to another CFTR co-chaperone CHIP [35]. Further, Aha1 increases CFTR trafficking to the cell surface in monolayer cultures by GSNO. While Aha1 knockdown with siRNA increases CFTR expression, it does not increase CFTR expression as much as knockout of another CFTR co-chaperone CHIP [35, 36]. However, the effect of Aha1 knockdown is magnified when SNOs are introduced. We also found that GSNO decreased Aha1 expression and diminished protein ATPase activity at 10 µM GSNO treatments resulting in lowered CFTR-Aha1 interaction. Taken together, the beneficial effect of SNOs in CFTR maturation is related in part to suppressing Aha1 expression and function. To our knowledge, this is the first demonstration that the increase in F508del CFTR maturation by SNOs involves the co-chaperone Aha1. SNOs may be noteworthy beyond their role in CFTR trafficking Aha1 associates with CFTR co-chaperones, which are also S-nitrosylated, and regulates their expression and function [19, 20].
Conclusion
Our study demonstrates that the interaction between SNOs and the co-chaperone Aha1 prevents F508del CFTR degradation. Our data suggest that S-nitrosothiol replacement has the potential to be an effective F508del CFTR corrector therapy. The success of ivacaftor (VX-770) in treating CF patients containing G551D mutations highlights the significance of seeking new approaches aimed at targeting other CFTR mutations, particularly F508del CFTR in airway epithelial cells. Although new corrector drugs developed for F508del CFTR represent major advances, they are not completely effective.
Conflicts of Interest: The authors declare that they have no conflicts of interest.
Acknowledgements
We thank Dr. Eric Sorscher from the, University of Alabama at Birmingham for providing CFBE41o- cell. Dr. John Riordan from the University of North Carolina for providing the monoclonal anti-CFTR antibody and Dr. Jeffrey L Brodsky from the University of Pittsburgh for valuable suggestions. This work was supported by the National Institute of Health 1PO1871-01A1.
References
-
Boucher RC (2002) An overview of the pathogenesis of cystic fibrosis lung disease. Adv Drug Delivery Rev 54(11): 1359-1371.
-
Riordan J (2008) CFTR function and prospects for therapy. Annu Rev Biochem 77: 701-726.
-
Kopito RR (1999) Biosynthesis and degradation of CFTR. Physiol Rev 79(1): S167-S172.
-
Grove DE, Rosser MF, Ren HY, Naren AP, Cyr DM (2009) Mechanisms for rescue of correctable folding defects in CFTR ΔF508. Mol Biol Cell 20(18): 4059-4069.
-
Sawczak V, Getsy P, Zaidi A, Sun F, Zaman K, et al. (2015) Novel approaches for potential therapy of cystic fibrosis. Current Drug Targets 16(9): 923-936.
-
Gaston B, Doctor A, Singel D, Stamler JS (2006) S-Nitrosothiol signaling in respiratory biology. Am J Respir Crit Care Med 173(11): 1186-1193.
-
Lancaster J, Gaston B (2004) NO and nitrosothiols: spatial confinement and free diffusion Am J Physiol Lung Cell Mol Physiol 287(3): 465-466.
-
Gaston B (1999) Nitric oxide and thiol groups. Biochim Biophys Acta 1411(2-3): 323-333.
-
Grasemann H Gaston B, Fang K, Paul K Ratjen F (1999) Decreased levels of nitrosothiols in the lower airways of patients with cystic fibrosis and normal pulmonary function. J Pediatr 135(6): 770-772.
-
Palmer LA, Gaston B Johns RJ (2000) Normoxic stabilization of hypoxia-inducible factor-1 and activity:Redox-dependent effect of nitrogen oxides. Mol Pharmacol 58(6): 1197-1203.
-
Gaston B, Reilly J, Drazen JM, Fackler J, Ramden P, et al. (1993) Endogenous nitrogen oxides and bronchodilator Snitrosothiols in human airways. Proc Natl Acad Sci USA 90(23): 10957-10961.
-
Li D, Shirakami G, Zhan X, Johns R (2000) Regulation of ciliary beat frequency by the nitric oxide-cyclic guanosine monophosphate signaling pathway in the rat airway epithelial cells. Am J Respir Cell Mol Biol 23(2): 175-181.
-
Jain L, Chen XJ, Brown LA, Eaton DC (1998) Nitric oxide inhibits lung sodium transport though a cGMP-mediated inhibition on epithelial cation channels. Am J Physiol 274(4): 475-484.
-
Kamosinska B, Radomski MW, Duszyk M, Radomski A, Man SF (1997) Nitric oxide activates chloride currents in human lung epithelial cells. Am J Physiol 272(6): 1098- 1104.
-
Persichini T, Colasanti M, Gauro G, Ascenzi P (1998) Cysteine nitrosylation inactivates the HIV-1 protease. Biochem Biophys Res Commun 250(3): 575-576.
-
Saura M, Zaragoza C, McMillanA, Hohenadl C, Lowenstein JM, et al. (1999) An antiviral mechanism of nitric oxide: Inhibition of a viral protease. Immunology 10(1): 21-26.
-
Zaman K, Bennett D, Butler M, Greenberg Z, Getsy P, et al. (2014) S-nitrosoglutathione diethyl ester increases cystic fibrosis transmembrane regulator expression and maturation in the cell surface. Biochem Biophys Res Commun 443(4): 257-262.
-
Zaman K, Palmer LA, Doctor A, Hunt J, Gaston B (2004) Concentration-dependent effects of endogenous S-nitrosoglutathione on gene regulation by specificity proteins Sp3 and Sp1. Biochem J 380: 67-74.
-
Marozkina N, Yemen S, Borowitz M, Liu L, Plapp M, et al. (2010) Hsp70/Hsp90 organizing protein as a nitrosylation target in cystic fibrosis. Proc Natl Acad Sci USA 107(25): 11393 -11398_._
-
Zaman K, Carraro S, Doherty J, Henderson E, Lendermon E, et al. (2006) A novel class of compounds that increase CFTR expression and maturation in epithelial cells. Mol Pharmacol 98(3): 1435-1442.
-
Zaman K, McPherson M, Vaughan J, Hunt J, Mendes F, et al. (2001) S-Nitrosoglutathione increases cystic fibrosis transmembrane regulator maturation. Biochem Biophys Res Commun 284(1): 65-70.
-
Snyder A, McPherson M, Hunt JF, Stamler JS, Gaston B (2002) Acute effects of aerosolized S-nitrosoglutathione in cystic fibrosis. Am J Respir Crit Care Med 165(7): 1-5.
-
Chen L, Patel RP, Teng X, Bosworth CA, Lancaster JR (2006) Mechanisms of cystic fibrosis transmembrane conductance regulator activation by S-nitrosoglutathione. J Biol Chem 281: 9190-9199.
-
Howard M, Fischer H, Roux J, Santos B, Gullans S, Yancey P, Welch P (2003) Mammalian osmolytes and S-Nitrosoglutathione promote F508 CFTR protein maturation and function. J Biol Chem 278(37): 35159- 35167.
-
Andersson C, Gaston B, Roomans G (2002) S-Nitrosoglutathione induces functional ∆F508 CFTR in cultured airway epithelial cells. Biochem Biophys Res Commun 297(3): 552-557.
-
Servetnyk Z, Krujkova J, Gaston B, Zaman K, Hjelte L, et al. (2006) Activation of delF508 CFTR in CF airway epithelial cell lines and CF nasal epithelial cells by S-nitrosoglutathione. Respir Res 7: 124-130.
-
Sun F, Mi Z, Condliffe SB, Bertrand CA, Gong X, et al. (2008) Chaperone displacement from mutant cystic fibrosis transmembrane conductance regulator restores its function in human airway epithelia. FASEB J 22(99): 3255-3263.
-
Varga K, Goldstein R, Jurkuvena A, Chen L, Matalon S, et al. (2008) Enhanced cell-surface stability of rescued ΔF508 cystic fibrosis transmembrane conductance regulator (CFTR) by pharmacological chaperonce. Biochem J 410(3): 555-564.
-
Okiyoneda T, Barriere H, Bagdany M, Rabeh WM, Du K, et al. (2010) Poripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 329(5993): 805-810.
-
Brodsky J, Frizzell R (2015) A combination therapy for cystic fibrosis. Cell 163(1): 17.
-
Lukacs G, Verkman A (2012) CFTR: folding, misfolding and correcting the ΔF508 conformational defect. Tends Mol Med 18(2): 81-91.
-
Wang X, Venable J, LaPointe P, Hutt D, Koulov A, et al. (2006) Hsp90 co-chaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 127(4): 803-815.
-
Ihrig V, Obermann W (2017) Identifying inhibitors of the Hsp90-Aha protein complex, a potential target to drug cystic fibrosis. SLAS Diccov 22(7): 923-928.
-
Zaman K, Sawczak V, Zaidi A, Butler M, Zeinomar M, et al. (2016) Augmentation of CFTR maturation by S-nitrosoglutathione reductase in epithelial cells. Am J Physiol Lung Cell Mol Physiol 310(3): L263-L270.
-
Zaman K, Knight J, Hussain F, Cao R, Estabrooks S, et al. (2019)S-nitrosylation of CHIP enhances F508del CFTR maturation. Am J Resp Cell Mol Biol 61(6): 765-775.
-
Addy C, Schock B (2019) Putting CHIP (s) on the table: Introducing Nitrosothiols into the arena of CFTR modulation. Am J Resp Cell Mol Biol 61(6): 673-675.
- Superposition of Cryo-EM and AlphaFold Predictions of Dengue Antigen-Antibody Complexes
- Jugular-Applied Coherent Low-Level Laser Therapy Enhances Systemic Mitochondrial Metabolic Function and Antioxidant Response
- Role of OMC32 Polypeptide in Acrosin-Mediated Exocytosis during the Bovine Sperm Acrosome Reaction
- Association of Galectin-3 but not Laminin in Tamoxifen-Induced Growth Suppression in Breast Cancer MCF-7 Cells
- Effect of Different Wavelengths of Light on the Rate of Photosynthesis
- Nutritional, Therapeutic, and Environmental Effect of Oyster Mushrooms: An Editorial