Vogt-Koyanagi-Harada Disease: Placoid-Like Presentation Associated with Serous Retinal Detachment
Background: Vogt-Koyanagi-Harada disease is a multisystemic autoimmune disorder that is characterized by bilateral, chronic and granulomatous panuveitis. A placoid-like presentation of this condition is unusual and it can mimic other causes, such as syphilis. Complete evaluation and early treatment might improve visual outcome and decrease number of recurrent episodes. Case Presentation: We report a case of a 21 year-old female patient with progressive low visual acuity in both eyes and a placoid-like lesion associated with serous retinal detachment in the fundus exam. Optical coherence tomography showed a highly reflective subretinal membranous structure and subretinal fluid. Fluorescein angiography showed multiple areas of pinpoint leakage and optic disk staining. Other ancillary exams, such as serology and head/orbit computerized tomography were all normal. Cerebrospinal fluid revealed pleocytosis. The diagnosis of Vogt-Koyanagi-Harada was made and prompt treatment with high-dose systemic corticosteroid was started. The patient had a complete recovery of her visual acuity and has had no recurrence of inflammation so far. Conclusion: This article describes an unusual presentation of Vogt-Koyanagi-Harada disease evaluated with multimodal imaging and also highlights the benefits of the early diagnosis and prompt management
Introduction
Vogt-Koyanagi-Harada (VKH) disease is a multisystemic autoimmune disorder with ophthalmic, auditory, integumentary and neurologic manifestations [1, 2]. The main ocular finding of this disease is bilateral, chronic and granulomatous panuveitis [1].
VKH disease most frequently occurs in the adult population and women are more affected than men [2]. In Brazil, VKH disease is the most common cause of noninfectious panuveitis [3]. VKH disease usually manifests in four clinical stages: prodromal, acute uveitic, convalescent and chronic recurrent. The prodromal phase is characterized by neurologic and auditory symptoms, such as headache, neck stiffness, tinnitus and hearing loss. The acute uveitic phase shows diffuse choroiditis with serous retinal detachment and optic disk swelling. “Sunset glow fundus” due to the depigmentation of choroid is a hallmark of the convalescent phase. Recurrent episodes of anterior uveitis are characteristic of the chronic recurrent phase [2].
According to the Revised Diagnostic Criteria, VKH disease can be classified as complete, incomplete or probable. Complete disease is characterized by the involvement of ocular, intergumentary, auditory/neurology systems. Incomplete disease is an ocular disease with either integumentary or auditory/neurology manifestation. Probable VKH is an uveitis consistent with VKH without any extra ocular manifestation [2, 4].
Fluorescein angiography (FA) is a helpful method in the evaluation and diagnosis of VKH disease [5]. This exam shows pinpoint leakage, large placoid areas of hyperfluorescence, pooling of the dye within the subretinal fluid and optic nerve staining [5, 6]. Optical coherence tomography (OCT) is another method with increasing importance in the evaluation of this condition. Subretinal membranous structure, serous retinal detachment, subretinal hyperreflective dots and choroidal thickening have been described by many authors in VKH disease [3, 5, 7].
Multiple treatment regimens have been established for VKH disease, such as intravenous, oral and topical corticosteroid, cyclosporine, azathioprine, mycophenolate mofetil, cyclophosphamide and infliximab [2]. Early high- dose systemic corticosteroid followed by slow tapering over a minimum of 3 to 6 months are mandatory to suppress the inflammation in the acute phase [2]. Most cases of recurrence are due to the inadequate treatment during initial-onset acute VKH disease [2, 3]. Visual prognosis worsens if the inflammation is prolonged or therapy is delayed [5, 8].
The objective of this article is to describe an unusual presentation of this condition evaluated with multimodal imaging and also to highlight the benefits of the early diagnosis and prompt management.
Case Report
We report a case of a 21 year-old black female who presented to our emergency room in Hospital Sao Paulo (Escola Paulista de Medicina, Federal University of Sao Paulo, Sao Paulo, Brazil) with a chief complaint of progressive low visual acuity in the right eye for the past 3 weeks and in the left eye for the past 1 week. The onset of ocular symptoms was associated with headache. She denied any other symptoms, history of trauma or ocular surgery. Her past medical and family history was all unremarkable. She was using topical dexamethasone in both eyes every 6 hours prescribed by the ophthalmologist in the city that she lives (Belo Horizonte- Minas Gerais, Brazil) due to the diagnosis of anterior uveitis. Her initial ophthalmological exam revealed a best-corrected visual acuity of 20/40 in the right eye and count fingers at 1m in the left eye. The slit-lamp examination showed cells and flare of a mild grade in the anterior chamber without any other notable findings. Her intraocular pressure (IOP) was 12 mmHg in both eyes.
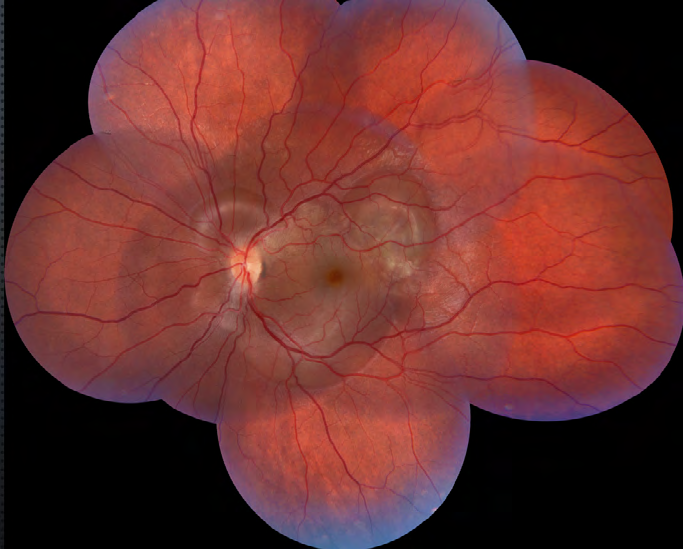
The fundus examination of the right eye revealed a pink optic disk with blurred margins, a whitish round intraretinal lesion in the temporal inferior vascular arcade and a yellowish placoid-like lesion temporal to the fovea (Figure 1). Further, we observed a serous macular detachment in the papillomacular bundle and around the optic disk. In the left eye, the fundus examination showed a pink optic disk, with blurred margins and multiple serous retinal detachment in the macula (Figure 2).
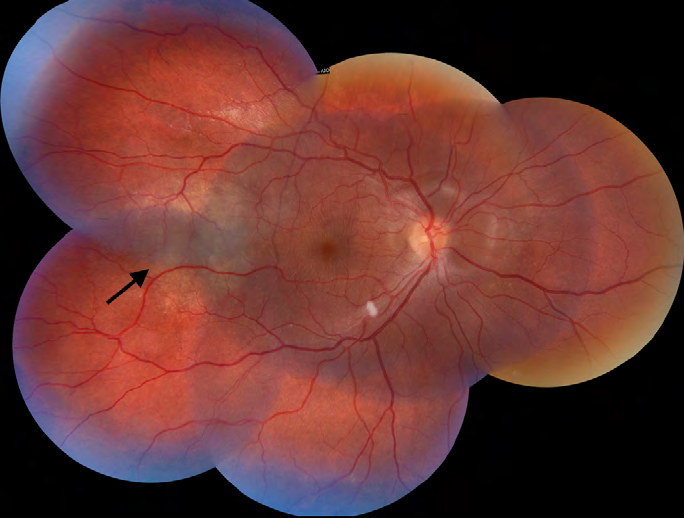
Ancillary examination performed at the emergency room included a clinical evaluation, laboratory work-up, serology, computerized tomography (CT) of head/orbit and a lumbar puncture. Her arterial blood pressure was 100/70 mmHg, the blood count, erythrocyte sedimentation rate, C-reactive protein levels, renal and hepatic function were within normal range values. The results of blood samples ruled out toxoplasmosis, syphilis and HIV. The tuberculin skin test measured 4 mm and the head/orbit CT scan did not detect any abnormalities. The cerebrospinal fluid evaluation revealed pleocytosis (cellularity of 880 with 86% of lymphocytes).
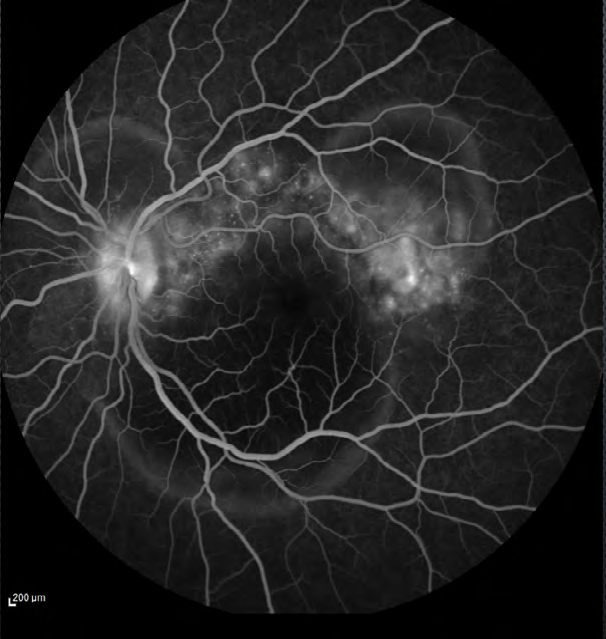
The ophthalmological ancillary examination, such as color fundus photography, OCT, FA and ocular ultrasound were also performed. FA showed multiple areas of pinpoint leakage and staining of the optic nerve in both eyes (Figure
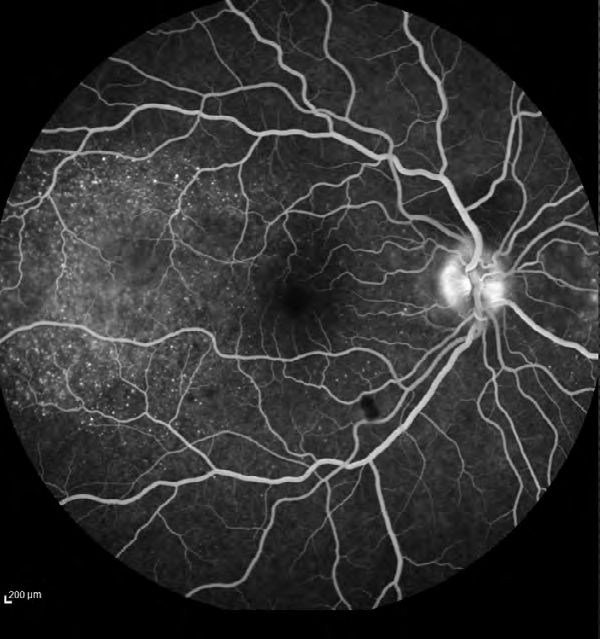
3). OCT of the macular region of the right eye showed serous retinal detachment in the papillomacular bundle and around the optic disk with a subretinal hyperreflective structure temporal to the fovea and bacillary layer detachment near the optic disk (Figure 4). OCT of the macular region of the left eye showed multiple serous retinal detachment (Figure 5). There were few posterior vitreous cells in both eyes. The ocular ultrasound revealed a serous retinal detachment in the posterior pole, diffuse thickening of the posterior wall, without signs of posterior scleritis.
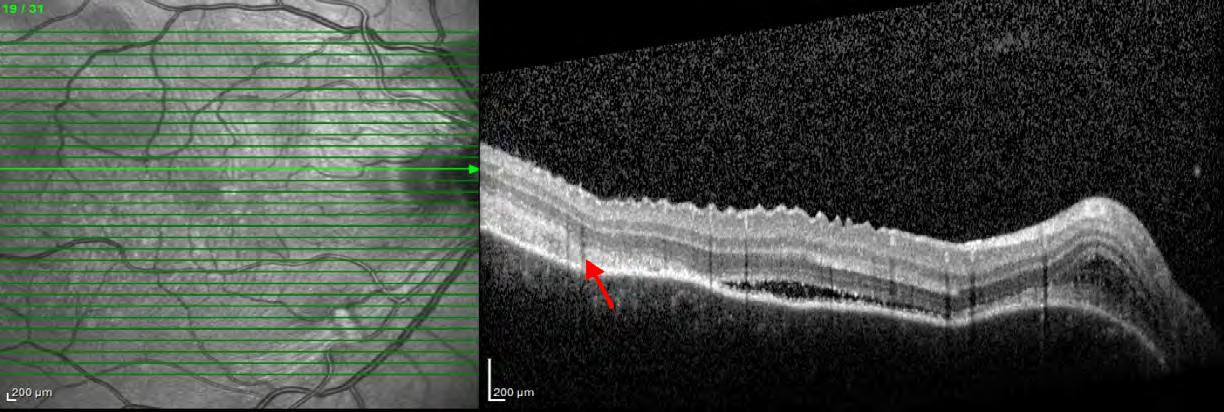

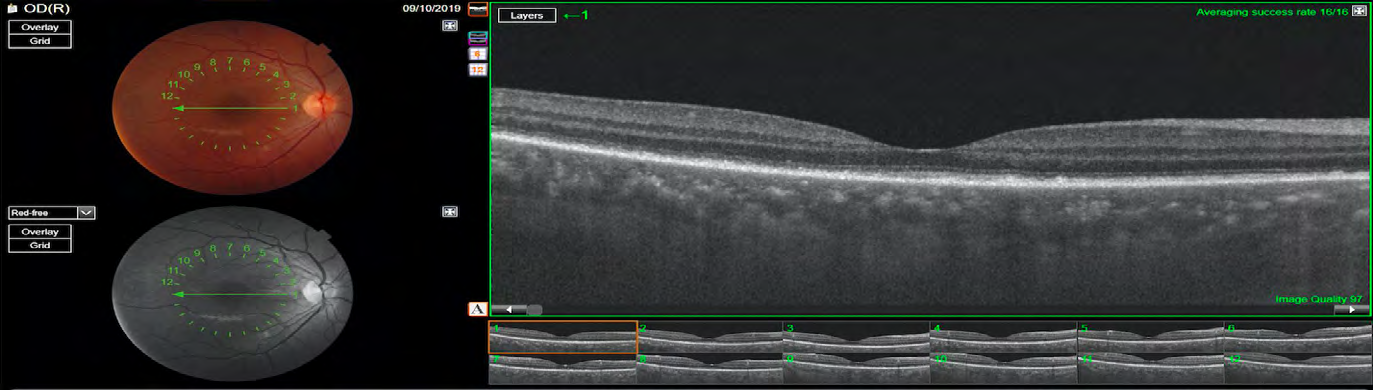
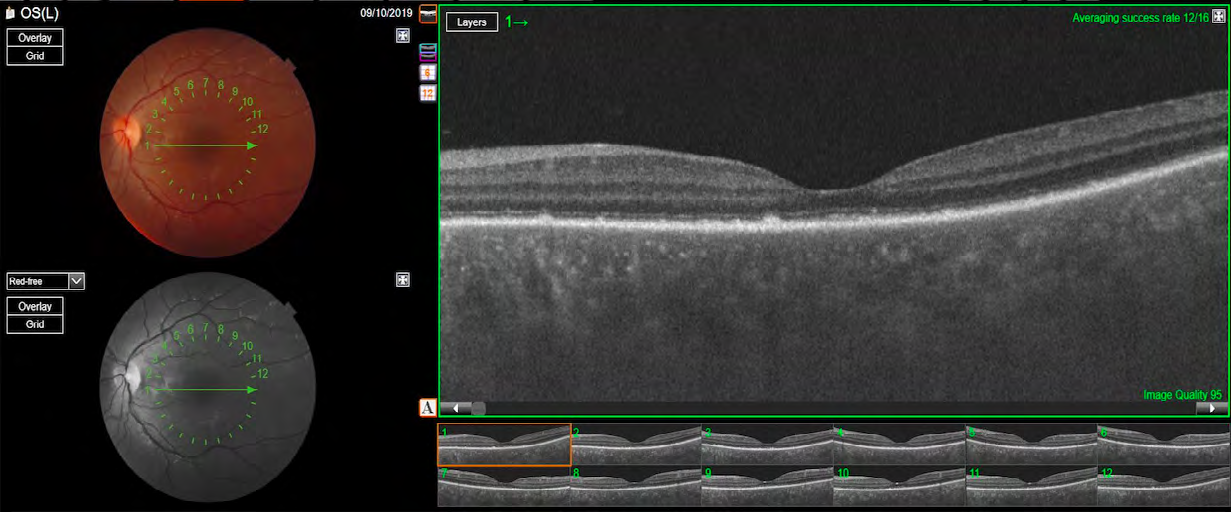
After this complete evaluation, the diagnostic most likely was incomplete VKH disease according to the Revised Diagnostic Criteria for VKH disease. At this point, the patient started the treatment with high-dose intravenous corticosteroid (1g methylprednisolone per day for 3 days). The intravenous corticotherapy was converted to high-dose oral corticosteroid treatment (prednisone 1mg/kg/day), following with a slow tapering. Within 10 days after the start of the treatment, her visual acuity improved to 20/25 OD and 20/50 OS, the IOP did not increase and the serous retinal detachment reduced. She was using 50 mg per day of prednisone. At the 4 month follow-up visit, her visual acuity was 20/20 in both eyes, the IOP was 14 mmHg in both eyes and the fundus exam revealed complete improvement of the serous retinal detachment. OCT showed some hyperreflective irregularities at the level of RPE/Bruch membrane complex, with no subretinal fluid and resolution of the hyperreflective subretinal structure. (Figure 6 & 7). She was no longer using any of corticosteroid or immunosuppressive treatment.
Discussion and Conclusion
VKH disease is an important cause of noninfectious panuveitis in Brazil and it can lead to several complications that include cataract, glaucoma and choroidal neovascular membrane (CNV), which, ultimately, may also lead to visual impairment [9].
This case shows the importance of the early diagnosis and prompt management of VKH disease. In Brazil, it is important to rule out infectious causes that can mimic this presentation, such as syphilis. Prompt diagnosis and treatment with corticosteroid with slow tapering may result in good visual recovery and reduced number of recurrent episodes [2].
Many authors have described a membranous structure on OCT, that appeared as a highly reflective line in the subretinal space that represents a portion of outer photoreceptor segment that becomes separated from the inner segment layer by cystoid space [3, 5, 8, 9].
It was discussed that this membranous form of the outer segment was associated with deposit of inflammatory material such as fibrin. After the starting therapy with corticosteroid the membranous structure might change to a granular structure [5].
VKH with a placoid-like presentation was firstly described by Wright et al. 8 patients presented widespread serous retinal detachment accompanying the multifocal pigment epithelial lesions and associated with uveitis, papilitis and systemic manifestations such as headache and tinnitus. The appearance of during initial transit of fluorescein angiography was identical to that in acute posterior multifocal placoid pigment epitheliopathy (APMPPE) with early hypofluorescence of the lesion and late staining. Evaluation with OCT of the placoid lesions was not performed [10].
In our case, we described a yellowish placoid lesion temporal to the fovea with a serous retinal detachment adjacent. The FA revealed multiple pinpoint leakages in that location and the OCT showed a subretinal highly reflective structure with no cystoid space. We believe that finding represents a dense deposit of inflammatory debris, such as fibrin, that resolved after the treatment.
References
-
Huang G, Peng J, Ye Z, Kijlstra A, Zhang D, et al. (2018) Multispectral image analysis in Vogt-Koyanagi-Harada disease. Acta Ophthalmol 96(4): 411-419.
-
Lodhi SAK, Lokabhi Reddy JM, Peram V (2017) Clinical spectrum and management options in Vogt-Koyanagi- Harada disease. Clin Ophthalmol 11: 1399-1406.
-
Pichi F, Invernizzi A, Tucker WR, Munk MR (2020) Optical coherence tomography diagnostic signs in posterior uveitis. Prog Retin Eye Res 75: 100797.
-
Read RW, Holland GN, Rao NA, Tabbara KF, Ohno S, et al. (2001) Revised diagnostic criteria for Vogt-Koyanagi- Harada disease: Report of an international committee on nomenclature. Am J Ophthalmol 131(5): 647-652.
-
Liu XY, Peng XY, Wang S, You QS, Li YB, et al. (2016) Features of Optical Coherence Tomography for the Diagnosis of Vogt-Koyanagi-Harada Disease. Retina 36(11): 2116-2123.
-
Rao NA, Gupta A, Dustin L, Chee SP, Okada AA, et al. (2010) Frequency of Distinguishing Clinical Features in Vogt-Koyanagi-Harada Disease. Ophthalmology 117(3): 591-599.
-
Manethova K, Ernest J, Hrevus M (2017) Vogt- Koyanagi-Harada syndrome (uveomeningoencephalitic syndrome). Eur J Ophthalmol 27(1): 5-8.
-
Vasconcelos-Santos D V, Sohn EH, Sadda S, Rao NA (2010) Retinal pigment epithelial changes in chronic Vogt- Koyanagi-Harada disease: Fundus autofluorescence and spectral domain-optical coherence tomography findings. Retina 30(1): 33-41.
-
Read RW, Rechodouni A, Butani N, Johnston R, LaBree LD, et al. (2001) Complications and prognostic factors in Vogt-Koyanagi-Harada disease. Am J Ophthalmol 131(5): 599-606.
-
Wright BE, Bird AC, Hamilton AM (1978) Placoid pigment epitheliopathy and Harada’s disease. Br J Ophthalmol 62(9): 609-621.
- Screening of Hospital Staff During World Glaucoma Week in a Tertiary Eye Care Centre
- Angioid Streaks with Macular Neovascularization: Clinical Insights from Two Cases
- Giant Kissing Naevus: An Oculoplastic Challenge
- Why Freedom of Vision Should Not Cost the Freedom of Feeling - LASIK in the Climate of Change
- Asymmetric Optic Nerve with Small Disc and Large Cup: A Rare and Challenging Case of Unilateral Optic Nerve Hypoplasia
- Large Angle Exotropia in a Child: A Case Report