Comparative Modeling and Molecular Interaction Study for the Management of AMD and CRVO Ocular Disorder
Age Related Macular Degeneration (AMD) and Central Retinal Vein Occlusion (CRVO) are the rare and leading cause of blindness among patients with ocular problem. Many proteins are reported in the progression of these ocular disorders. Proteins which are directly involved in the development of this disorder reported in the literature, their sequence related information retrieved from biological databases. In silico technique was implemented in order to characterize the properties and structures of the proteins using ProtParam. For studying about the potential phosphorylation sites in protein generally NetPhos server was used whereas for denoting the location of signal peptide cleavage sites and their presence the server which is used is SingalP server. For prediction of secondary structure prediction of proteins is done by using SOPMA. The SOSUI server performs the identification of trans-membrane regions. The 3D dimensional structure was modeled using Swiss Model Workspace and Modeller. Ramachandran plot was used to validate the stereochemical properties of the predicted structures because it is a very important step after 3D structure prediction. Docking of screened phytochemicals with selected proteins was performed by AutoDock. Docking study revealed that Curcumin (binding energy: -8.35) and Berberine (binding energy: -7.14) can be used as better therapeutic lead molecule for the cure of CRVO and AMD respectively
Introduction
Age-related macular degeneration (AMD) is deterioration/breakdown of the eye’s macula. AMD results in a loss of vision in the center of the visual fields due to the damage in the retina [1]. Age-related macular degeneration shows with characteristic yellow deposits i.e. dursen in the macular region [2]. Hemicentin1, extracellular matrix protein that is expressed specifically in their retinal pigmented epithelial cells. Complement Factor H which is glycoprotein that plays an integral role in a regulation of the complement mediated immune system, complex processing and programmed cell death, Vascular Endothelial Growth Factor A. Generally, blood flows into the retinal part passing through the central retinal artery (CRA) and leaves out through the central retinal vein (CRV) [3]. Main reason due to which Central Retinal Vein Occlusion (CRVO) is caused is because of a blood clot in the CRV i.e. central retinal vein, Activated Protein C (APC) which is a serine protease that performs the blood clotting, Coagulation Factor V is a protein of coagulation system which plays a role as a co- factor, Activated Factor VII, the thrombophilia factor and Antithrombin III, protein molecule in plasma that inactivates thrombin are the specific proteins responsible for causing CRVO.
Computational packages and online servers are the current tools used into the protein sequence analysis and characterization [4]. As the physiochemical characterization of proteins provides the better idea about the properties such as atomic composition, molecular weight, isoelectric points, aliphatic index, GRAVY, instability index, extinction effect. These parameter plays role in apprehending properties of protein analysis.
Prediction of protein secondary structure is also some other critical parameter in structural and practical evaluation of it. The major idea is to version the shape of Hemicentin 1 and Coagulation Factor V (protein of unknown structure) based at the template of a sequence homolog of acknowledged structure. Docking studies provide distinctive view of drug-receptor interplay. In the course of work, virtual screening was done by applying docking studies by AutoDock 4.0. Resveratrol, Luteolin and Berberine are the phytochemicals responsible for treatment of AMD whose docking performed and Curcumin and Alpha lipoic acid are the phytochemicals responsible for treatment of CRVO whose docking was performed.
Materials and Methodology
Flow Chart
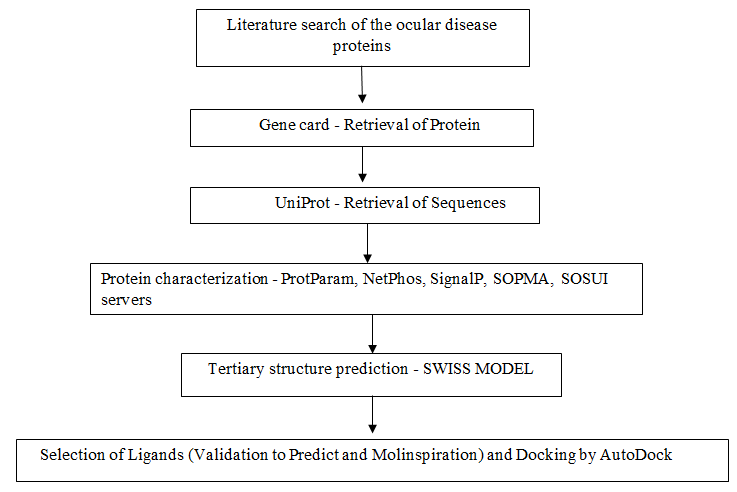
Protein Sequence Retrieval
The protein sequences were retrieved from the manually curated public protein database UNIPROT [5]. The seek end result yielded 7 ocular protein sequences of the illnesses AMD and CRVO with the aid of random choice and feature prepared a non-redundant records set (Table 1). The protein sequences have been retrieved in FASTA file format and used for evaluation.
| Accession number | Sequence description | Organism |
|---|---|---|
| Q96RW7 | Hemicentin 1 | Homo Sapiens (Human) |
| P08603 | Complement factor H | Homo Sapiens (Human) |
| P15692 | Vascular endothelial growth factor A (VEGFA) | Homo Sapiens (Human) |
| P06681 | Activated protein C | Homo Sapiens (Human) |
| P12259 | Coagulation factor 5 | Homo Sapiens (Human) |
| P08709 | Coagulation factor 7 | Homo Sapiens (Human) |
| P01008 | Antithrombin III | Homo Sapiens (Human) |
Table 1: Protein sequences retrieved from Swiss-Prot database.
Physio-Chemical Characterization
The physio-chemical parameters, theoretical isoelectric point, molecular weight, total quantity of superb and terrible residues, extinction coefficient [6], half-life [7, 8, 9, 10], instability index [11], aliphatic index [12] and grand common hydropathy (GRAVY)[13] have been computed the usage of the Expasy’s ProtParam prediction server. The NetPhos 2.0 server used in studying the potential phosphorylation sites of the protein [14]. Further, SignalP 4.1 server used to denote the location and presence of the single peptide cleavage sites in given sequences [14].
Secondary Structure Prediction
The secondary structure prediction was done by using SOPMA server [15].
Molecular Modeling
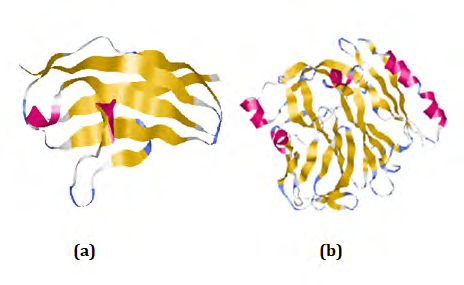
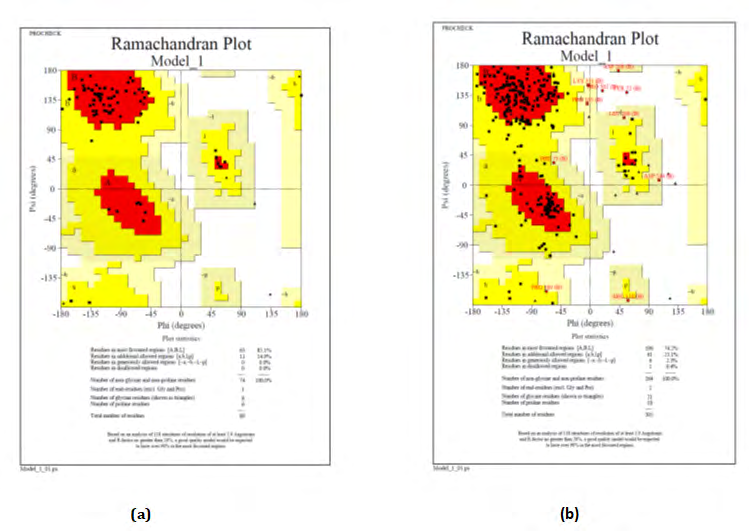
The tertiary structure prediction was done by using SWISS-MODEL. SWISS-MODEL is a structural bioinformatics web server dedicated to homology modeling of protein 3D structures. Nowadays, it consist of 3 main components that are: The SWISS-MODEL pipeline, The SWISS-MODEL Workshop, The SWISS-MODEL Repository. PDBsum is a database which provides a glance overview of contents of each 3D macromolecular structures present in PDB. It shows the molecule that make up the structure and schematic diagrams of their interactions.
Pocket Identification
Identification of geometric properties of protein pockets which are assumable position on protein surface was performed by using CASTp [16].
Ligand Selection/Validation
The ligands were selected by using Molinspiration tool by calculating partition coefficient (logP), Molecular polar surface area (TPSA) and molecular volume. It also pay attention to Lipinski’s rule of 5. In order to find that the selected ligand is toxic or non-toxic, ToxPredict tool have been used [17].
Docking
Docking is probably the best known computational methods used to identify the fit between a receptor and a potential ligand. Ligand is selected through literature search for this study. The 3D structure of Hemicentin 1 and Coagulation factor 5 are docked by using Autodock4.0 for virtual screening. The goal of docking is to predict the binding affinity and the bound conformation [18].
Results and Discussion
The physicochemical parameters, theoretical isoelectric point, molecular weight, total number of positive and negative residues, extinction coefficient, instability index, aliphatic index and grand average hydropathy (GRAVY) were computed using the Expasy’s ProtParam prediction server and tabulated in Table 2.
| Accession no. | Sequence length | M.wt | pI | -R | +R | EC | II | AI | GRAVY |
|---|---|---|---|---|---|---|---|---|---|
| Q96RW7 | 5,635 | 79282.7 | 6.07 | 77 | 70 | 74760 | 36.31 | 92.4 | -0.068 |
| P08603 | 1,231 | 61230.1 | 6.44 | 65 | 62 | 121890 | 38.74 | 56.69 | -0.601 |
| P15692 | 232 | 27042.3 | 9.21 | 24 | 40 | 39055 | 52.3 | 57.54 | -0.783 |
| P06681 | 752 | 83267.8 | 7.23 | 77 | 77 | 96840 | 40.87 | 78.68 | -0.298 |
| P12259 | 2,224 | 82282.9 | 5.83 | 93 | 77 | 126795 | 41.95 | 72.42 | -0.5 |
| P08709 | 466 | 51593.8 | 6.92 | 51 | 50 | 69005 | 48.68 | 79.27 | -0.288 |
| P01008 | 464 | 52602.4 | 6.32 | 62 | 60 | 45880 | 39.48 | 84.68 | -0.278 |
Table 2: Physico-chemical Parameters computed using Expasy’s ProtParam tool.
The NetPhos2.0 server was used for studying potential phosphorylation sites of protein (Figure 2a-2g).
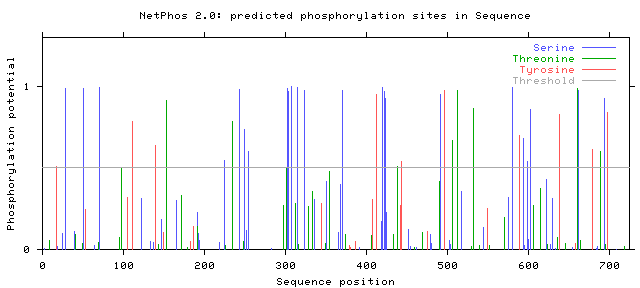
Figure 2a: Results of NetPhos analysis: Hemicentin 1.
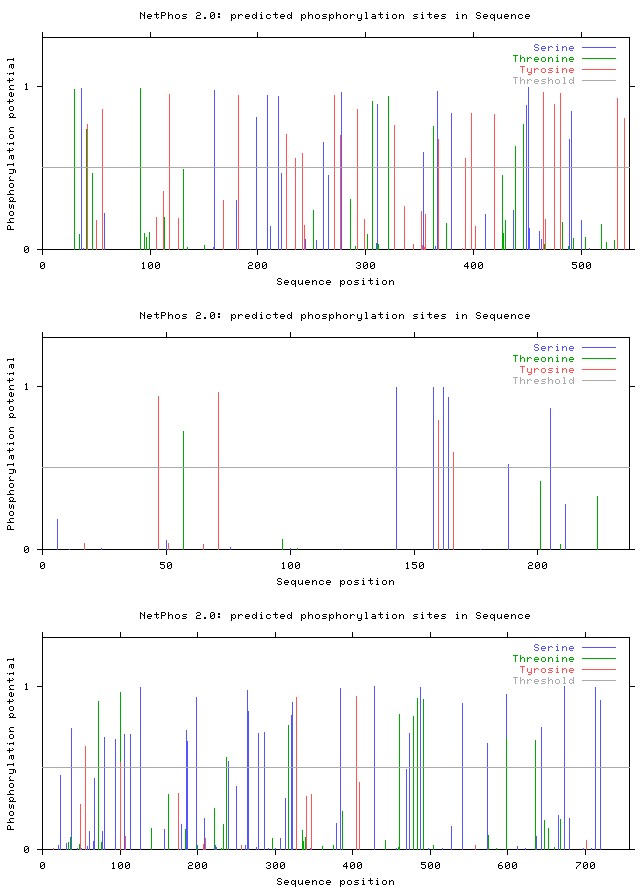
Figure 2b: Results of NetPhos analysis: Complement factor H.
Figure 2c: Results of NetPhos analysis: VEGF A.
Figure 2d: Results of NetPhos analysis: Activated protein C.
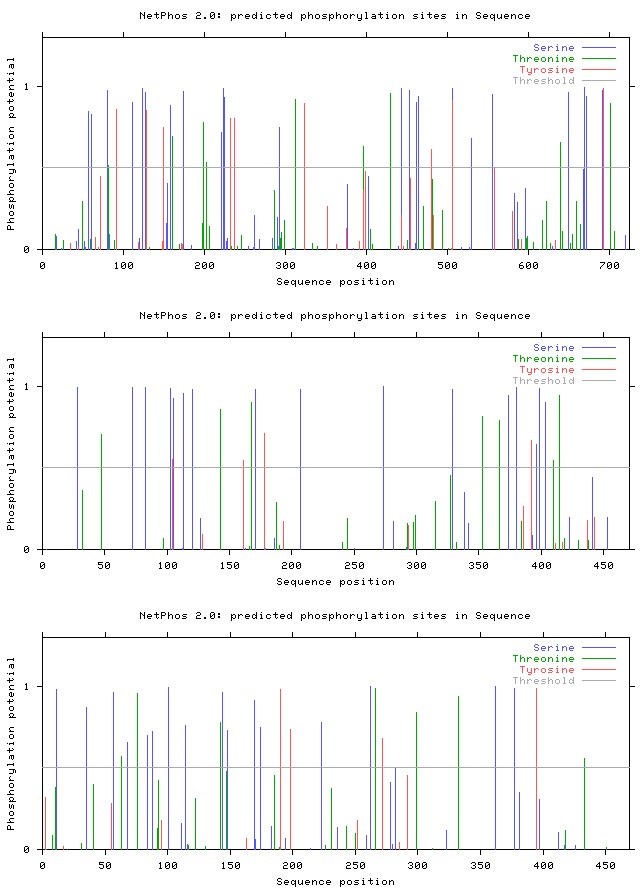
Figure 2e: Results of NetPhos analysis: Coagulation factor V.
Figure 2f: Results of NetPhos analysis: Coagulation factor VII.
Figure 2g: Results of NetPhos analysis: Antithrombin III.
SignalP4.1 server was used for denoting the presence and location of signal peptide cleavage sites in given sequences (Figures 3a- 3g).
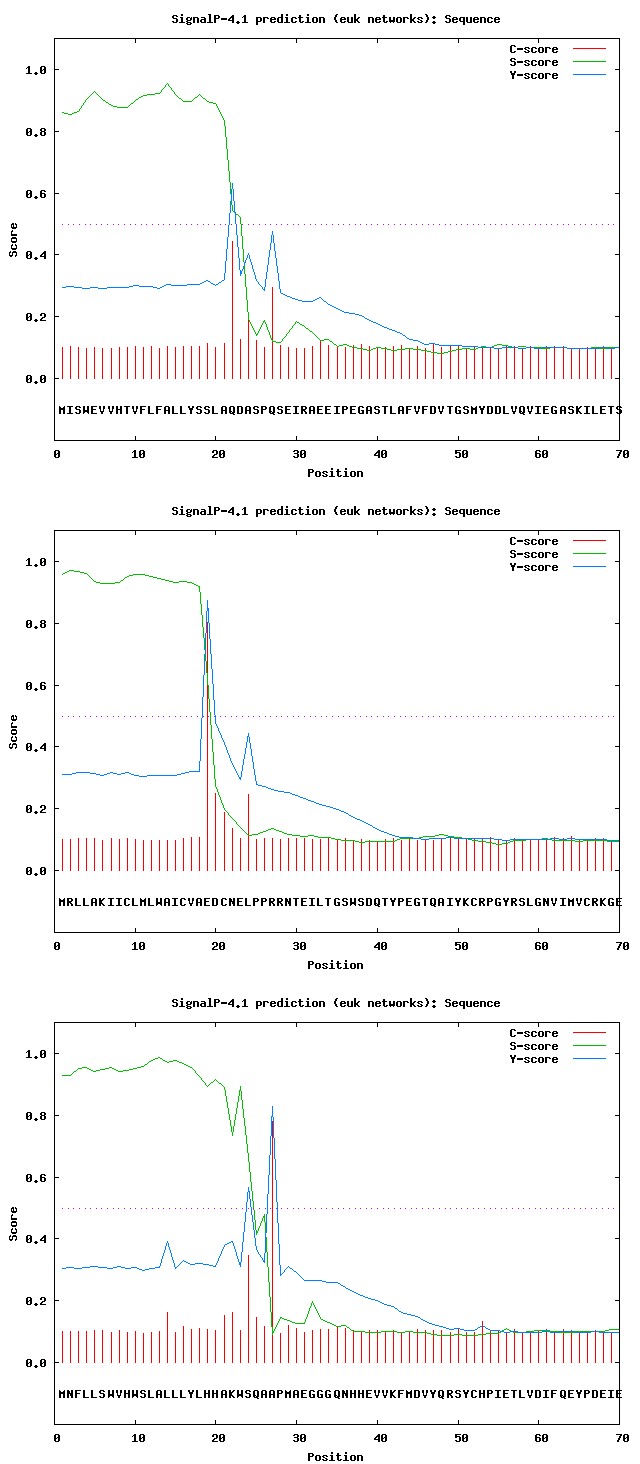
Figure 3a: Results of SignalP analysis: Hemicentin 1.
Figure 3b: Results of SignalP analysis: Complement factor H.
Figure 3c: Results of SignalP analysis: VEGF A.
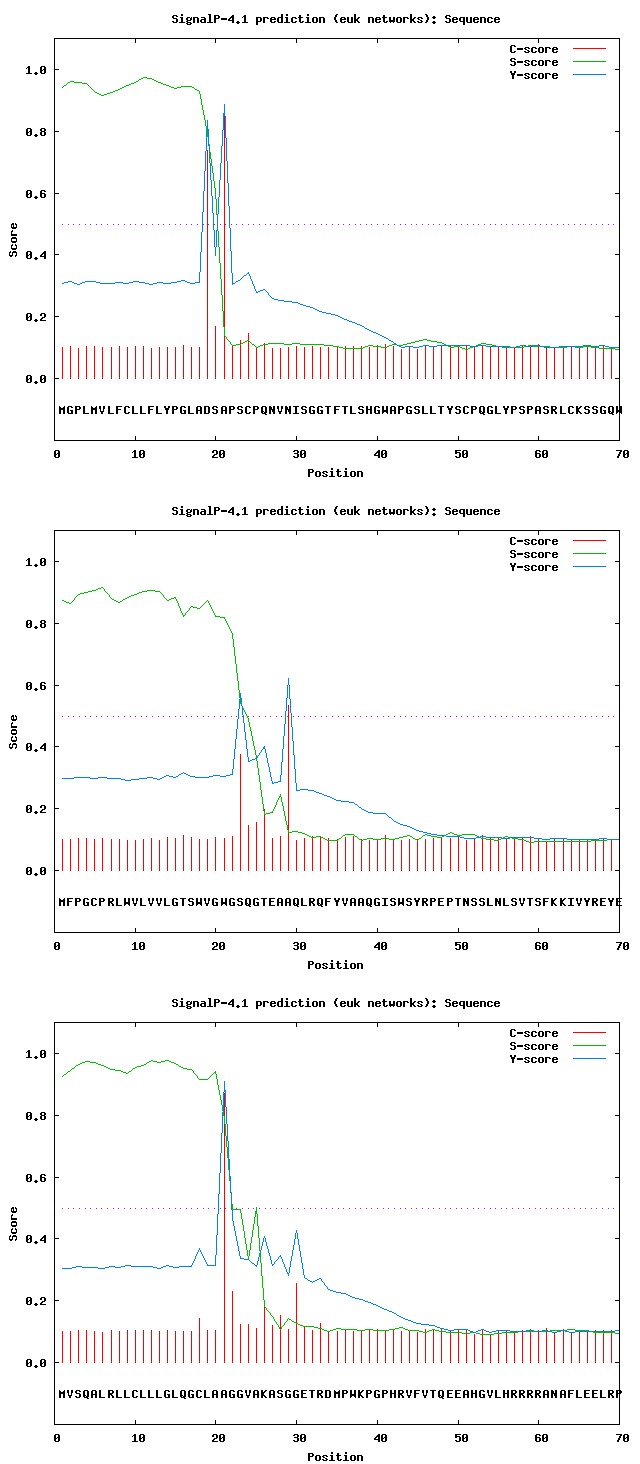
Figure 3d: Results of SignalP analysis: Activated protein C.
Figure 3e: Results of SignalP analysis: Coagulation factor V.
Figure 3f: Results of SignalP analysis: Coagulation factor VII.
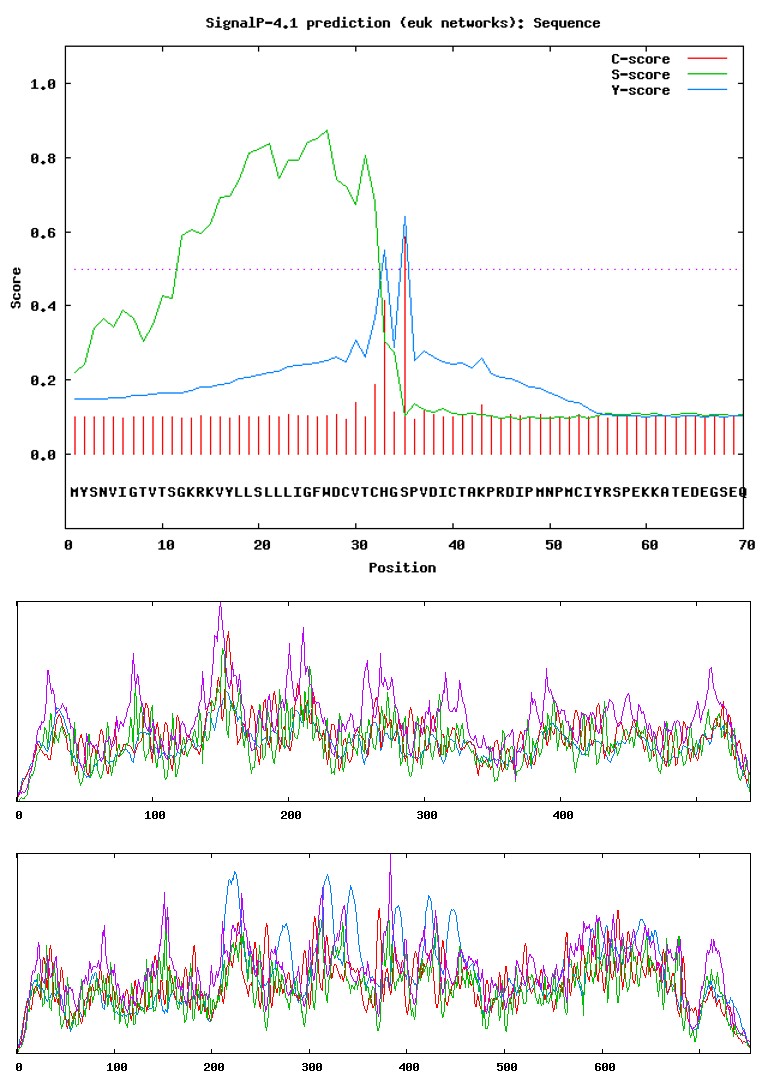
Figure 3g: Results of SignalP analysis: Antithrombin III.
The tool SOPMA was used for the secondary structure prediction of proteins (Table 3) (Figure 4a-4e)).
| Accession Number | Alpha helix | 310 helix | Pi helix | Beta bridge | Extended strand | Beta turn | Random coil | Ambiguous state | Other states |
|---|---|---|---|---|---|---|---|---|---|
| Q96RW7 | 17.6 | 0 | 0 | 0 | 30.14 | 5.56 | 46.94 | 0 | 0 |
| P08603 | 2.41 | 0 | 0 | 0 | 20.37 | 6.67 | 70.56 | 0 | 0 |
| P15692 | 22.84 | 0 | 0 | 0 | 12.5 | 1.72 | 62.93 | 0 | 0 |
| P06681 | 26.99 | 0 | 0 | 0 | 17.82 | 4.39 | 50.8 | 0 | 0 |
| P12259 | 15.42 | 0 | 0 | 0 | 25.14 | 7.22 | 52.22 | 0 | 0 |
| P08709 | 26.18 | 0 | 0 | 0 | 18.67 | 7.51 | 47.64 | 0 | 0 |
| P01008 | 39.22 | 0 | 0 | 0 | 17.03 | 5.17 | 38.58 | 0 | 0 |
Table 3: Secondary parameters computed using SOPMA server.
Figure 4a: Results of SignalP analysis: Hemicentin 1.
Figure 4b: Results of SignalP analysis: Complement factor H.
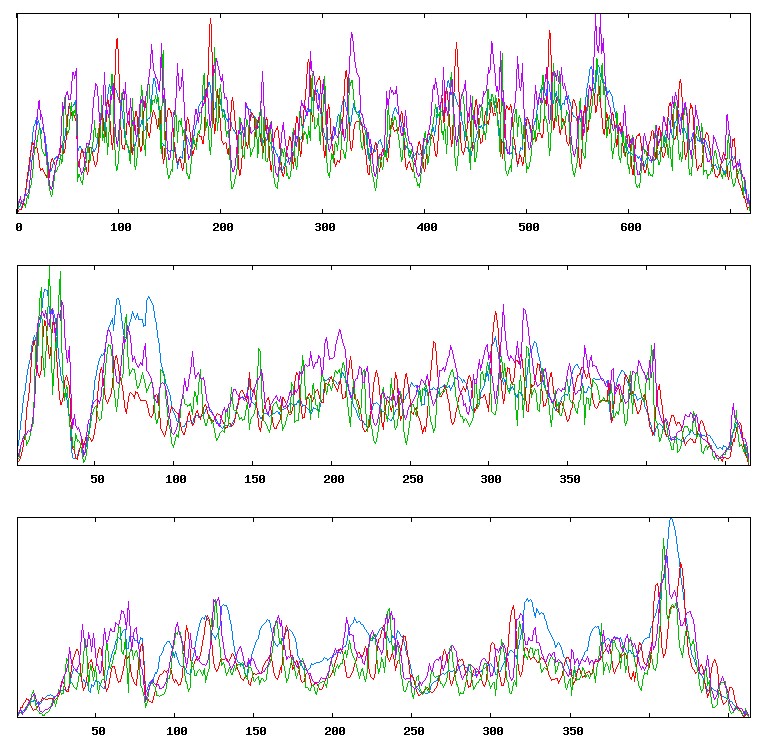
Figure 4c: Results of SignalP analysis: VEGF A.
Figure 4d: Results of SignalP analysis: Activated protein C.
Figure 4e: Results of SignalP analysis: Coagulation factor V.
The SOSUI server performed the identification of transmembrane regions (Table 4a, 4b) (Figure 5a, 5b).
| No. | N terminal | transmembrane region | C terminal | type | length |
|---|---|---|---|---|---|
| 1 | 1 | MISWEVVHTVFLFALLYSSLAQD | 23 | PRIMARY | 23 |
| 2 | 699 | IEAPKLMVVQSELLVALGDITV | 720 | PRIMARY | 22 |
Table 4: Screened herbal molecules for protein ligand docking studies.
Table 4a: Transmembrane regions using SOSUI server: Hemicentin 1.
| No. | N terminal | transmembrane region | C terminal | type | length |
|---|---|---|---|---|---|
| 1 | 1 | MRLLAKIICLMLWAICVAEDCNE | 23 | PRIMARY | 23 |
Table 5: Screened herbal molecules for protein ligand docking studies.
Table 4b: Transmembrane regions using SOSUI server: Complement factor H.
The screened herbal molecules (phytochemicals) for protein ligand docking studies are given below (Tables 5a, 5b) (Figures 6a, 6b).
| S.No. | Phytochemicals | PubChem ID | Chemical Formula | Molecular weight | Structure |
|---|---|---|---|---|---|
| 1 | Curcumin | 969516 | C21 H20 O6 | 368.379 | |
| 2 | Alpha lipoic acid | 864 | C8 H14 O2 S2 | 206.325 | |
| 3 | Berberine | 2353 | C20 H18 NO4+ | 336.361 | |
| 4 | Luteolin | 5280445 | C15 H10 O6 | 286.236 | |
| 5 | Resveratrol | 445154 | C14 H12 O3 | 228.243 |
Table 6: Screened herbal molecules for protein ligand docking studies.
The ligands were selected by using Molinspiration tool (Table 6).
| Compound | milogP | TPSA | MW | nON | nOHNH | nviol | nrotb | natoms | Volume |
|---|---|---|---|---|---|---|---|---|---|
| Curcumin | 2.303 | 93.066 | 368.379 | 6 | 2 | 0 | 8 | 27 | 332.182 |
| Alpha lipoic acid | 2.254 | 37.299 | 206.332 | 2 | 1 | 0 | 5 | 12 | 182.69 |
| Berberine | 0.196 | 40.821 | 336.367 | 5 | 0 | 0 | 2 | 25 | 296.302 |
| Luteolin | 1.974 | 111.123 | 286.239 | 6 | 4 | 0 | 1 | 21 | 232.067 |
| Resveratrol | 2.986 | 60.684 | 228.247 | 3 | 3 | 0 | 2 | 17 | 206.922 |
Table 7: ligands were selected by using Molinspiration tool.
Standard Rate Chart
TPSA 40-140, logP -4 - +5, MW<500 Dalton, n OHNH<5, n ON<10
Binding energy calculation results of drug receptor interaction, for different herbal compounds are given in Table 7.
| Protein | Phytochemical | Binding Energy | Reference RMSD |
|---|---|---|---|
| Coagulation factor V | Curcumin | -8.35 | 44.76 |
| Coagulation factor V | Alpha lipoic acid | -5.17 | 45.78 |
| Hemicentin 1 | Berberine | -7.14 | 40.23 |
| Hemicentin 1 | Luteolin | -6.52 | 40.42 |
| Hemicentin 1 | Resveratrol | -6.06 | 41.61 |
Table 8: Result of drug receptor interaction.
Results are illustrated in Figures 7a-7c showing the interaction of Hemicentin 1 protein domain with berberine, luteolin and resveratrol.
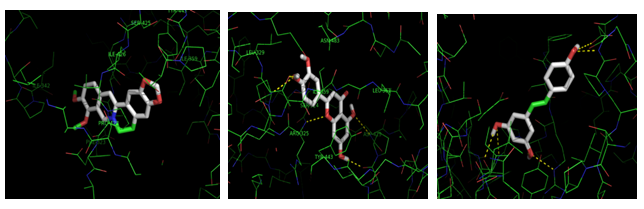
Results are illustrated in Figures 8a, 8b showing the interaction of Coagulation factor V protein domain with curcumin and alpha lipoic acid.

Conclusion
Insilico characterization is crucial in interpreting the crucial physical and chemical houses together with the prediction of fundamental affirmation of protein of their secondary systems. Many simple to advance features of proteins can give a main concept approximately their structural and useful factors. Furthermore assessment of outcomes in the course of Insilico characterization of multiple proteins gives very clean cut comparative consequences and aspects. In cutting-edge direction of work through evaluating the effects of selected proteins, physic-chemical characterization research provide a very good idea about the houses together with pi, EC, AI, GRAVY and instability index that are important and crucial in offering facts about the proteins and their residences. In the procedure of modeling, Hemicentin 1 and coagulation aspect V, notwithstanding the absence of homologous structures from structural databases, we have been capable of become aware of useful templates that percentage low sequence similarity with each proteins which, while blended together, embody the complete period of Hemicentin 1 and coagulation element V. In continuation to the study, herbal molecules were screened out through docking studies.
Screening of ligands is totally based on herbal molecules selections. Concluding the final selection of herbal molecules, Curcumin responsible for the treatment of CRVO is screened and Berberine responsible for the treatment of AMD is screened and validated as it shows best ligand properties after all sets of validation. Substantial study between Hemicentin 1 and coagulation factor V and natural ligands was analyzed to recommend more and more proficient search for potential target molecule against Central Retinal Vein Occlusion and Age Related Macular Degeneration disorders. Dexamethasone, Triamcinolone and Minocycline are the commercial drugs available in the market for the treatment of CRVO. Bevacizumab, Lucentis, Fenretinide and Ranibizumab are the commercial drugs available in the market for the treatment of AMD. Thus, the results will be a fruitful opportunity for the development of new herbal drug against these idiopathic disorders. Virtual
screening for potential ligand can give new insights towards the therapeutic intonations and alterations towards the advances in treatment for Central Retinal Vein Occlusion and Age Related Macular Degeneration disorders.
References
-
National Eye Institute (2015).
-
GBD 2015 Disease and Injury Incidence and Prevalence Collaborators (2016).
-
Olver J, Cassidy L (2005) Ophthalmology at a Glance. 1st(Edn.), Blackwell Science, Oxford, England, pp: 112.
-
K Sivakumar (2005) Adv Biotech IV 27.
-
Boeckmann B, Bairoch A, Apweile R, Blatter MC, Estreicher A, et al. (2003) The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucl Aciac Res 31(1): 365-370.
-
Bachmair A, Finley D, Varshavsky A (1986) _In-Vivo_ Half Life of a Protein Is a Function of Its Amino Terminal Residue. Science 234(4773): 179-186.
-
Gonda DK, Bachmair A, Wünning I, Tobias JW, Lane WS, et al. (1989) Universality and Structure of the N-end Rule. J Biol Chem 264(68): 16700-16712.
-
Tobias JW, Shraden TE, Rocap G, Varshavsky A (1991) The N-End Rule in Bacteria. Science 254(5036): 1374- 1377.
-
Ciechanover A, Schwartz AL (1989) How are substrates recognized by the ubiquitin-mediated proteolytic system. Trends Biochem Sci 14(12): 483-488.
-
Guruprasad K, Reddy BVB, Pandit MW (1990) Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting _in-vivo_ stability of a protein from its primary sequence. Prot Eng 4(2): 155-161.
-
Ikai A (1980) Thermostability and Aliphatic Index of Globular Proteins. J Biochem 88(6): 1895-1898.
-
Kytte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157(1): 105-132.
-
Blom N, Gammeltoft S, Brunak S (1999) Sequence and structure based prediction of eukaryotic protein phosphorylation sites. Journal of Molecular Biology 294(5): 1351-1362.
-
Petersen TN, Brunak S, Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nature Methods 8: 785-786.
-
Geourjon C, Deleage G (1995) SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci 11(6): 681-684.
-
Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y (2006) CASTp: computed atas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucl Acids Res 34: W116-W118.
-
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23(1-3): 3-25.
-
Sousa SF, Fernandes PA, Ramos MJ (2006) Protein-ligand docking: Current status and future challenges. Proteins Structure Function and Bioinformatics 65(1): 15-26.
- Screening of Hospital Staff During World Glaucoma Week in a Tertiary Eye Care Centre
- Angioid Streaks with Macular Neovascularization: Clinical Insights from Two Cases
- Giant Kissing Naevus: An Oculoplastic Challenge
- Why Freedom of Vision Should Not Cost the Freedom of Feeling - LASIK in the Climate of Change
- Asymmetric Optic Nerve with Small Disc and Large Cup: A Rare and Challenging Case of Unilateral Optic Nerve Hypoplasia
- Large Angle Exotropia in a Child: A Case Report