A Rare Case Report of Meckel-Gruber Syndrome (MGS)
Meckel-Gruber syndrome (MGS) is an autosomal recessive disorder characterized by occipital encephalocele, polycystic kidneys and variable other congenital malformations. As an autosomal recessive disorder, the risk of recurrence of Meckel Gruber syndrome is 25%. The diagnosis of MGS is usually suspected with early prenatal ultrasound showing typical findings. As the patient was referred at 34 weeks, it was a late diagnosis. We report on a patient with MGS diagnosed by antenatal ultrasound scan. Pregnancy was terminated at 34 weeks of gestation.
Introduction
Meckel-Gruber Syndrome (MGS) is a rare, genetically heterogeneous and lethal autosomal recessive disorder characterized by occipital encephalocele, postaxial polydactyly and bilateral dysplastic cystic kidneys. This disorder is hypothesized to be caused by defects in primary cilia. MGS can be associated with anomalies including cleft lip and palate, anopthalmia, micropthalmia, ductal plate malformation of the liver, cardiac and genital anomalies. The diagnosis of MGS is considered when antenatal ultrasound shows associated findings. We here report on a baby with MGS based on antenatal ultrasound findings.
Case Report
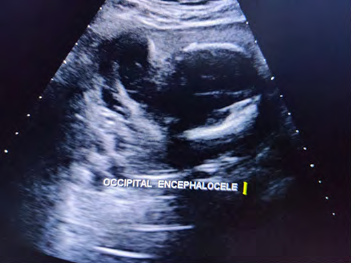
Twenty four years old G6 A5 presented at 34 weeks of gestation with a previous history of a baby with congenital malformations including Arnold Chiari. The parents are first degree cousins on both maternal and paternal sides. There was no other family history of congenital malformations. The mother denied taking any medications during pregnancy other than folic acid, which was started before pregnancy. Ultrasound scan, done at 15 weeks gestation stated as cystic hygroma. An ultrasound scan was repeated at 24 weeks gestation which confirmed posterior encephalocele and anhydramnios [1, 2].
Case was referred for detailed antenatal ultrasound at Niloufer hospital. On scanning, the findings were occipital encephaloele, polycystic kidneys, omphalocele, mesomelia [short limbs], bilateral talipes polydactyly, cleft lips and anhydramnios. The possibility of MGS was raised, the natural course and the prognosis was explained to the parents who opted to terminate the pregnancy [3]. Following further extensive genetic counseling and approval of the hospital ethics committee, termination of pregnancy was carried out at 34 weeks gestation. The baby was born alive but died in 5 minutes, no resuscitation was offered. Baby along with cord blood sample was sent for genetic analysis, results to be awaited. Karyotyping of both mother and father were normal (Figures 1-6).
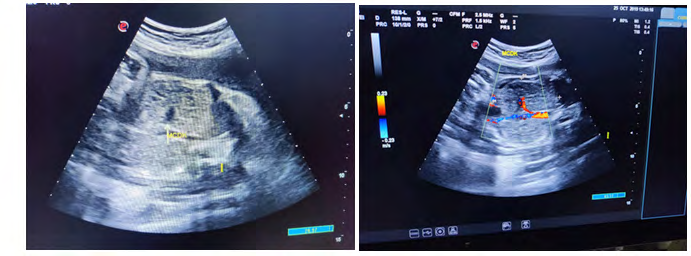
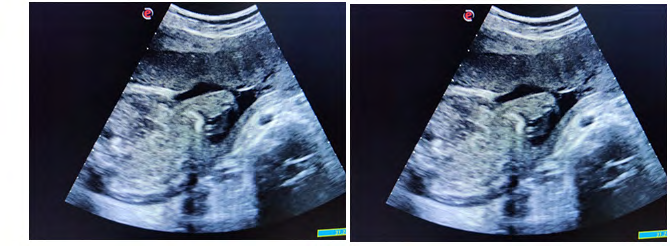
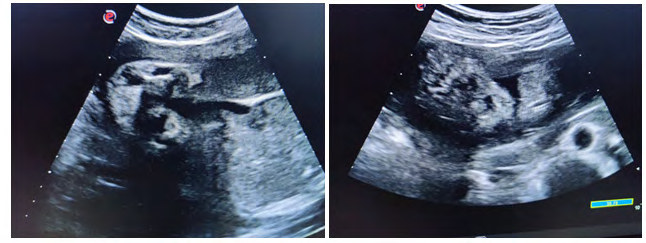
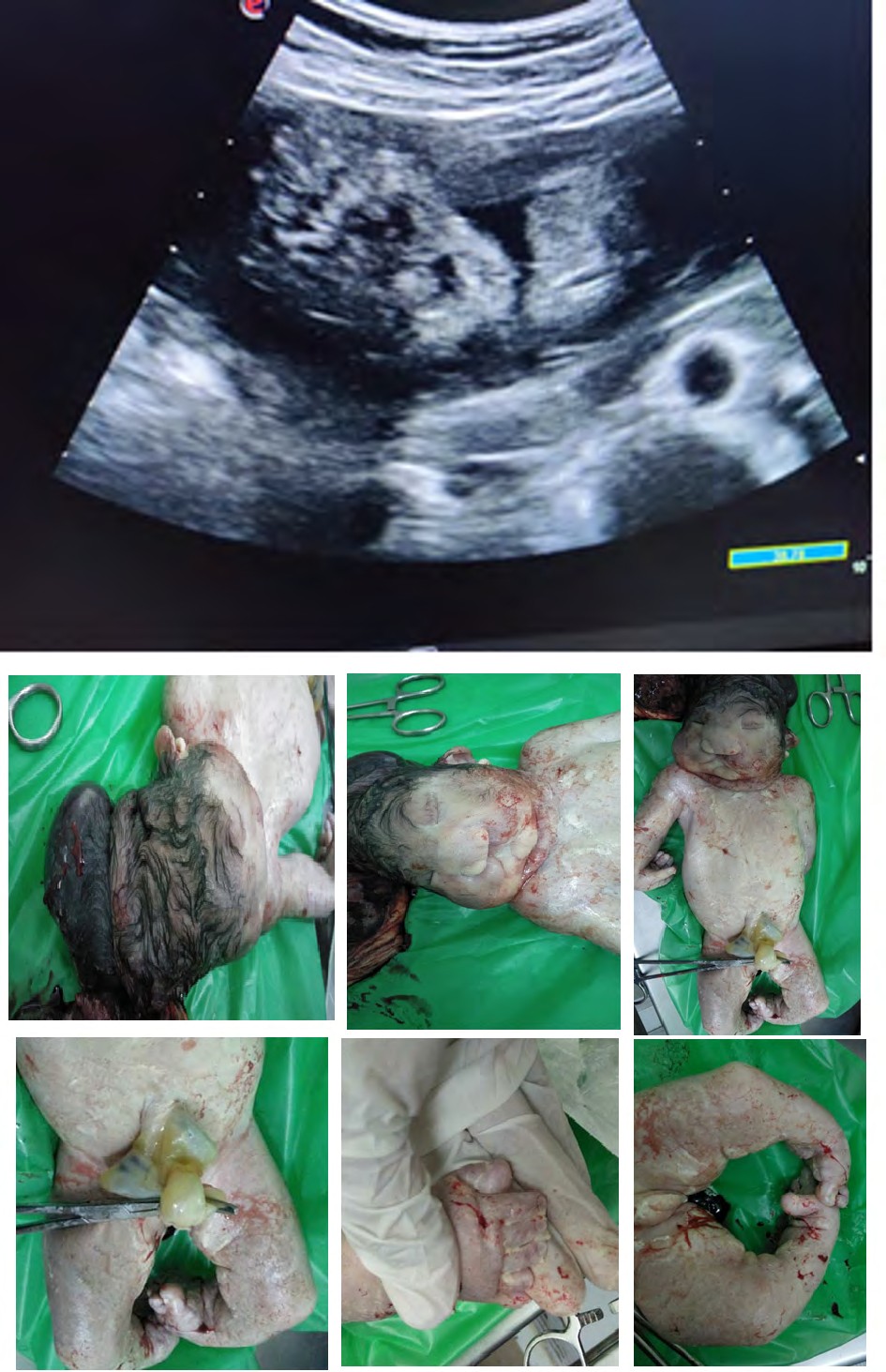
Figures 6: Pictures taken immediately after birth.
Discussion
Meckel-Gruber syndrome is a fatal [1], autosomal recessive disorder characterized by malformation of central nervous system, particularly occipital encephalocele, bilateral renal dysplasia, and polydactyly. As an autosomal recessive disorder, the risk of recurrence of Meckel Gruber syndrome is 25%. The diagnosis of MGS is usually suspected with early prenatal ultrasound showing typical findings. As the patient was referred at 34 weeks, it was a late diagnosis [4, 5, 6].
Conclusion
This case report highlights the importance of antenatal scan in diagnosis of inherited diseases such as MGS. Based on genetic analysis, we can suggest preimplantation genetic diagnosis (PGD) for the patient to prevent recurrence.
References
-
Meckel J (1882) Beschreibung zweier, durch sehr ähnliche Bildungsabweichungen entstellter Geschwister. Dtsch Arch Physiol 7: 99-172.
-
Opitz JM, Howe JJ (1969) The Meckel syndrome (dysencephalia splanchnocystica, the Grüber syndrome). Birth Defects Orig Art Ser 2: 167-179.
-
Salonen R, Opitz JM, Reynolds JF (1984) The Meckel syndrome-clinicopathological findings in 67 patients. Am J Med Genet 18(4): 671-689.
-
Salonen R, Paavola P (1998) Meckel syndrome. J Med Genet 35(6): 497-501.
-
Seppanen U, Herva R (1983) Roentgenologic features of the Meckel syndrome. Pediatr Radiol 13: 329-331.
-
Auber B, Burfeind P, Herold S, Schoner K, Simson G, et al. (2007) A disease causing deletion of 29 base pairs in intron 15 in the MKS1 gene is highly associated with the campomelic variant of the Meckel-Gruber syndrome. Clin Genet 72(5): 454-459.
- Ultrasound Guided Therapeutic Nerve Blocks
- Cyclops Lesion Without ACL Reconstruction: A Rare Case in a Patient with Intact Anterior Cruciate Ligament and Tibial Plateau Fracture
- Dosimetric Comparison between Two Dose Calculation Algorithms in SBRT Treatment of Lung Cancer in Ring-based and C-arm Radiation Therapy Equipment
- Adolescent Testicular Adrenal Rest Tumors: A Case Report and Review of the Literature
- Giant Intrathoracic Lipoma: A Rare Presentation
- Image of a Right Renal Angiomyolipoma Complicated by Hemorrhage