The E368Q Mutant Allele of GJA8 is Associated with Congenital Cataracts with Intrafamilial Variation in a South Indian Family
<p>Purpose: To determine the basis of the autosomal dominant congenital cataracts in a three generation south Indian pedigree. Methods: The proband and several family members underwent a complete ophthalmic examination. The coding regions of eight candidate genes (CRYAA, CRYBB2, CRYGC, CRYGD, GJA3, GJA8, AQP0, and PITX3) were amplified by PCR and directly sequenced. Wild type and mutant connexin50 (Cx50) were expressed by stable transfection of HeLa cells. Their cellular distributions and function were examined by Immunofluorescence microscopy and by microinjection of gap junction permeant tracers, respectively. Results: Congenital cataracts (with some variations in phenotype) segregated as an autosomal dominant trait within a three generation pedigree. Three affected individuals (proband, sibling and mother) showed a sequence variation in the candidate gene GJA8 encoding Cx50: a c.1102G>C transversion encoding a substitution of glutamate for glutamine at position 368 (E368Q). This substitution was absent from an unaffected family member (paternal aunt) and 100 healthy controls of the same ethnicity. In transfected HeLa cells, both wild type Cx50 and E368Q localized to gap junction plaques, and supported similar levels of intercellular transfer of Neurobiotin. Conclusions: The E368Q mutant allele of GJA8 is associated with autosomal dominant congenital cataracts with phenotypic variability. E368Q forms gap junction plaques and functional channels in transfected HeLa cells.</p>
Introduction
Congenital cataract is the leading cause of blindness in children [1, 2]. Its significance is particularly important in developing countries like India, where 7-15% of cases of childhood blindness are due to cataracts [3, 4, 5]. Cataracts may present either in isolation or as part of a metabolic disease or genetic syndrome. Up to 25% of isolated childhood cataracts are familial, typically with autosomal dominant inheritance and complete penetrance [6, 7]. These congenital cataracts vary widely on the basis of location, appearance and progression of opacity in the lens. According to Cat-map, an online chromosome map and reference database for inherited and age-related forms of cataracts, about 200 loci have been linked to inherited and age-related forms of human cataract [8]. However, a small number of genes account for most of the cases of inherited congenital cataracts [9]. About 50% result from mutations of α-, β-, and γ- crystallins, which are the major water soluble proteins of the lens. About 25% result from mutations of genes encoding the gap junction proteins (connexin46 (Cx46, GJA3) and connexin50 (Cx50, GAJ8)) or aquaporin 0 (MIP). The high prevalence of cataracts due to abnormalities of these major lens proteins is not surprising, since they are critical for maintaining the cellular structure, transparency and refractive properties of the lens. A variety of other gene mutations have also been identified (including other membrane proteins and other soluble proteins, like the transcription factor PITX3). In the present study, we have identified a novel mutant form of Cx50 by candidate gene screening of a family with inherited congenital cataracts. An initial characterization of its cellular and physiological behaviors is reported.
Materials and Methods
A prospective study of cases of childhood cataract registered at the Appasamy Krishna Hospital, Velachery, and Chennai, India was conducted in accordance with the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the Institutional Ethical Committee of Dr. ALM - Post Graduate Institute of Basic Medical Sciences, University of Madras, Chennai, and South India. A team of pediatric ophthalmologists serving at the hospital performed a clinical examination of the patients, and the senior ophthalmologist of the group confirmed the diagnosis. The ophthalmic examination of the anterior segment of the eye (including cornea, iris, and lens) was performed using slit-lamp biomicroscopy after pupil dilation in adult patients. Examination of the posterior segment of the fundus was done using a direct ophthalmoscope. Visual acuity was assessed by an optometrist using Snellen’s charts. Case histories were recorded using a questionnaire designed for the study. On securing informed consent, 5 ml of blood samples were collected from all the available members of the family. Genomic DNA was isolated from blood samples as previously described [10]. The coding exons of eight candidate genes (CRYAA, CRYBB2, CRYGC, CRYGD, GJA3, GJA8, PITX3 and MIP) were amplified from 50 ng of template genomic DNA in 60 µl reaction volumes [11]. PCR products were purified (BIO Basic Inc., NY, USA) and commercially sequenced (1st BASE Pte Ltd., Singapore). The sequences were checked for homology through NCBI- BLASTN analysis. Plasmid DNA encoding E368Q was obtained by PCR using Phusion DNA polymerase (New England BioLabs, Ipswich, MA), plasmid template containing wild type Cx50 in pcDNA 3.1/Hygro(+) (Invitrogen Life Technologies) and oligonucleotides encoding the nucleotide substitution (sense: 5’- GGGGAGAAAGTAGAGACCCCCGGAGTGGATAAGG-3’ and Antisense: 5’-CTGTGGCACGGCCACCTTCTCCTGCTCCTC-3’) using the strategy published by Minogue et al. [12]. HeLa cells stably expressing E368Q were obtained after transfection using Lipofectin® Transfection Reagent and PLUSTM Reagent (Invitrogen Life Technologies, USA) and selection in tissue culture medium containing 400 μg/ml hygromycin. HeLa cells stably expressing wild type Cx50 were produced previously [13] and were maintained in growth medium supplemented with 1 mg/ml Geneticin (Invitrogen Life Technologies). Immunofluorescence localization of wild type or mutant Cx50 was performed as previously described [13]. Intercellular communication was assessed 5 min. after microinjection of a solution containing 5% Lucifer yellow (Sigma) and 9% Neurobiotin (Vector Laboratories) into a single cell within a confluent monolayer [14]. Neurobiotin was detected using Cy3-streptavidin. The extent of intercellular transfer was determined by counting the number of cells surrounding the microinjected cell that contained Neurobiotin. Co-injection of Lucifer yellow was used to facilitate identification of the microinjected cell, because Cx50 channels allow very limited transfer of this tracer.
Results
Clinical evaluation
A 6 year-old girl presented to the hospital with a complaint of congenital cataract. Slit-lamp examination revealed a posterior cortical cataract in her left eye and a mature cortical cataract in her right eye. No other abnormalities were detected by ophthalmologic or general physical examination (specifically, ocular movements were full, conjunctivae were clear, and the anterior chambers looked normal). The child underwent surgery shortly afterwards for removal of the cataractous lenses and insertion of prostheses. A pedigree was ascertained from the family to determine the pattern of inheritance (Figure 1).
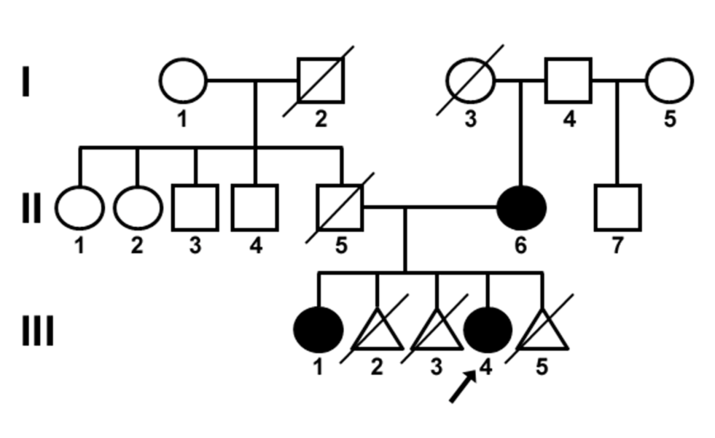
Figure 1: Pedigree of the family with autosomal dominant congenital cataracts. Black symbols indicate affected individuals and open symbols indicate unaffected individuals. Diagonal lines indicate deceased family members. The proband is indicated with an arrow. The proband (III-4), a sibling (III-1) and the mother (II-6) were affected. The other affected family members had different phenotypes than the proband: the mother (II-6) had posterior cortical cataracts in both eyes, and the proband’s elder sister (III-1) had blue dot cataracts in both eyes. There was no history of other ocular or systemic disorders within the family. The family pedigree suggested that the cataract developed spontaneously in the mother and followed an autosomal dominant inheritance pattern.
Molecular Characterization
Sequence analysis of all three affected members of the family (i.e., proband, sibling, and mother) revealed a c.1102G>C transversion in the coding region of the GJA8 gene (Figure 2).
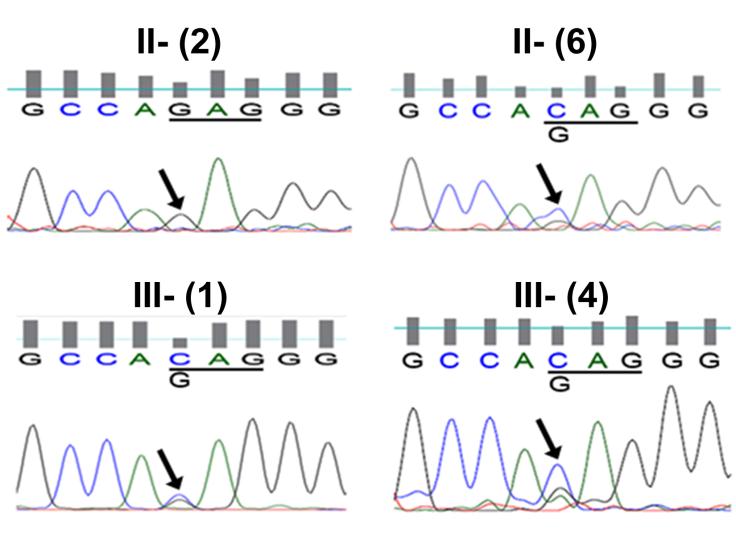
Figure 2: Chromatograms of DNA sequences show the association of a c.1102G>C transversion with the cataract phenotype in affected family members. The chromatogram from an unaffected individual (II-2) shows only the wild type GJA8 allele with G at position 1102. Sequence chromatograms from two affected family members (II-6 and III-1) and the proband (III- 4) show both C and G at position 1102. The mutant allele containing a G to C transversion would result in the substitution of glutamine for glutamate at amino acid residue 368 (E368Q). The seven other candidate genes did not show any pathogenic variations. The GJA8 transversion was not seen in an unaffected member of the family (paternal aunt, II-2). The mutation in GJA8 abolishes a restriction site for BspNCI. However, restriction site analysis using this enzyme could not be performed, because it is not commercially available. Therefore, all the control subjects (n = 100) were screened by direct sequencing of amplicons; the c.1102G>C transversion in GJA8 was not identified in any of them. Functional characterization The GJA8 gene encodes the gap junction protein Cx50 which forms intercellular channels in the lens. The c.1102G>C transversion would cause a substitution of glutamine for glutamate at position 368 (E368Q) in the cytoplasmic C-terminus of Cx50 (Figure 3).
![Figure 3: Diagram of the topology of Cx50 in the plasma membrane and the location of the E368Q mutation. The competence of E368Q to form gap junction plaques was assessed by localizing immunoreactive Cx50 in HeLa cells transfected with either wild type Cx50 or E368Q (Figure 4A,B). Wild type Cx50 and E368Q showed similar distributions. Both localized in the cytoplasm (possibly within the biosynthetic/secretory pathway) and at appositional membranes in a distribution consistent with gap junction plaques (arrows in Figure 4A, B). The abilities of wild type Cx50 and E368Q to support intercellular communication were assessed by studying the intercellular transfer of Neurobiotin from individual cells to their neighbors. Neurobiotin remained in the microinjected cell or transferred to very few neighbors in untransfected cells (Figure 4C). In contrast, Neurobiotin transferred to a significant number of cells in HeLa cells expressing Cx50 and E368Q. However, the extent of Neurobiotin transfer did not differ between wild type and mutant Cx50-expressing cells. Discussion In the present study, we have reported a family with a missense mutation in the Cx50 gene, GJA8, which is associated with congenital cataracts with intrafamilial variation. The mutation encodes a single amino acid substitution (E368Q) in the cytoplasmic C-terminus of the protein. The connexins are members of a family of proteins that form intercellular channels allowing the exchange of ions and small molecules between adjacent cells. A substantial number of mutations of the two human connexins that are expressed predominantly in lens fiber cells (Cx46 and Cx50) have been identified in families with inherited congenital cataracts [15]. In many cases,](/fulltextimages/51/fig_3.png)
Figure 3: Diagram of the topology of Cx50 in the plasma membrane and the location of the E368Q mutation. The competence of E368Q to form gap junction plaques was assessed by localizing immunoreactive Cx50 in HeLa cells transfected with either wild type Cx50 or E368Q (Figure 4A,B). Wild type Cx50 and E368Q showed similar distributions. Both localized in the cytoplasm (possibly within the biosynthetic/secretory pathway) and at appositional membranes in a distribution consistent with gap junction plaques (arrows in Figure 4A, B). The abilities of wild type Cx50 and E368Q to support intercellular communication were assessed by studying the intercellular transfer of Neurobiotin from individual cells to their neighbors. Neurobiotin remained in the microinjected cell or transferred to very few neighbors in untransfected cells (Figure 4C). In contrast, Neurobiotin transferred to a significant number of cells in HeLa cells expressing Cx50 and E368Q. However, the extent of Neurobiotin transfer did not differ between wild type and mutant Cx50-expressing cells. Discussion In the present study, we have reported a family with a missense mutation in the Cx50 gene, GJA8, which is associated with congenital cataracts with intrafamilial variation. The mutation encodes a single amino acid substitution (E368Q) in the cytoplasmic C-terminus of the protein. The connexins are members of a family of proteins that form intercellular channels allowing the exchange of ions and small molecules between adjacent cells. A substantial number of mutations of the two human connexins that are expressed predominantly in lens fiber cells (Cx46 and Cx50) have been identified in families with inherited congenital cataracts [15]. In many cases,
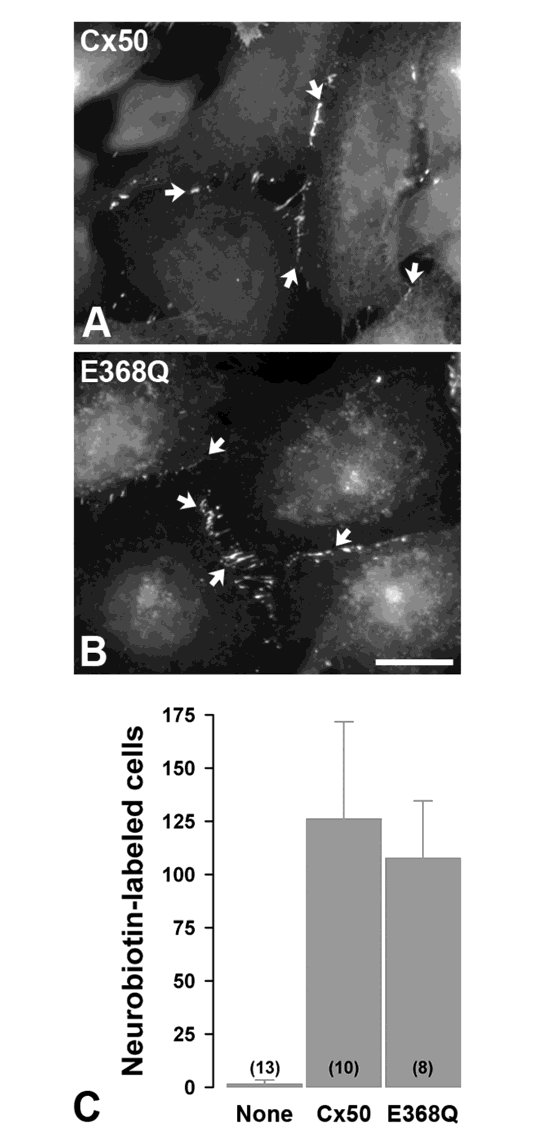
Figure 4: The E368Q mutant forms gap junction plaques and shows normal levels of intercellular communication. A, B. Immunofluorescence localization of wild type Cx50 (A) and E368Q (B) in stably transfected HeLa cells. Both wild type and mutant proteins show appositional membrane staining characteristic of gap junctions (arrows). Bar, 13 μm in A and 17 μm in B. C. Bar graphs show the extent of intercellular communication determined by counting Neurobiotin-labeled neighboring cells after microinjection of the tracer in untransfected HeLa cells (None) or cells expressing wild type Cx50 or E368Q. The numbers of microinjected cells are indicated in parentheses.
the mutation impairs gap junction function [15]. Studies of lens fiber cell connexin-null mice have demonstrated that connexin-based intercellular communication makes a contribution to the circulation of water and solutes that is critical to maintain lens transparency [16]. The cataract phenotype varied among family members in our study. Intrafamilial variations in the cataract phenotype are not without precedent. Members of a pedigree carrying the S73F Cx50 mutant have been previously reported to show opacities at different locations within the lens [17]. There is a wide spectrum of cataract types associated with different connexin mutants [15]; however, the relationship between genotype and phenotype is not understood. Indeed, the P88Q Cx50 mutation causes different phenotypes in different families [18, 19]. Moreover, the severity of the cataract caused by targeted deletion of Cx46 or Cx50 varies in different mouse strains, suggesting the influence of genetic background [20, 21]. The phenotypic differences in the cataract among members of our E368Q pedigree might similarly reflect differences in expression of modifying genes. The E368Q mutation occurs at the C-terminus of the connexin protein. Few pathologic substitutions of Cx46 or Cx50 have been identified within this domain. Two of the mutations identified in this region (Cx46fs380 and Cx50fs) are frame-shift mutations that cause cell biological abnormalities (cytoplasmic retrieval/retention or accelerated degradation) [12, 14]. Because these frame- shifts lead to replacement of many amino acids in the C- terminus of the connexin, the cellular and/or functional defects of these mutants may not bear much similarity with those of E368Q. One point substitution within the C- terminus of Cx50 (I247M) causes no significant functional abnormalities and has been suggested to be a polymorphism, not a pathological mutation [22]. In some experiments, deletion of segments of the Cx50 C-terminus does not interfere with formation of gap junction plaques or intercellular communication [14]. While a large truncation of the C-terminus can reduce function and alter pH-dependent gating [23, 24], the E368Q missense mutation is located more distal than the truncations studied in those reports. Therefore, it may not be surprising that we did not find abnormalities of gap junction formation or dye transfer in HeLa cells stably transfected with E368Q. The E368Q mutation might affect some non-channel functions of Cx50 such as facilitating cell-to-cell adhesion. However, this would not likely be altered in E368Q- expressing lens cells, since this mutant forms gap junction plaques normally. It is also possible that the E368Q mutation causes pathogenic abnormalities that would only be detectable in the lens. The C-terminal tail of Cx50 is subject to various post-translational modifications (e.g., phosphorylation and proteolytic cleavage) in the organ [25]. The E368Q mutation might affect protein conformation and alter accessibility to some of these sites. Unfortunately, analyses of the pathogenic mechanisms responsible for the E368Q cataract (and its phenotypic variability) are currently not feasible due to the unavailability of appropriate human tissue or of a corresponding animal model. In our assays, we did not identify abnormalities of gap junction formation or function for p.E368Q. This observation is consistent with its analysis using PolyPhen-2 which predicts that this is a benign substitution. The Exome Variant Server includes a different missense substitution at this site (c.1103A>G) resulting in p.E368G, which has not been studied, but is also predicted to be benign. Thus, c.1102G>C (p.E368Q) is a GJA8 (Cx50) allele that is rather rare in the general population and is associated with cataracts in at least one family. However, it may also be a benign variant.
Acknowledgments
This work was supported by National Institutes of Health grant EY08368 (to ECB) and through funding from UGC-SAP- DRS II and DST-FIST (GSK, KDK, STS).
References
-
World Health Organization. Prevention of Childhood Blindness. WHO, 1992.
-
Resnikoff S, Pascolini D, Etya'ale D, Kocur I, Pararajasegaram R, et al. (2004) Global data on visual impairment in the year 2002. Bull World Health Organ 82(11): 844-851.
-
Rahi JS, Dezateux C (2001) Measuring and interpreting the incidence of congenital ocular anomalies: lessons from a national study of congenital cataract in the UK. Invest Ophthalmol Vis Sci 42(7): 1444-1448.
-
Gilbert CE, Canovas R, Hagan M, Rao S, Foster A (1993) Causes of childhood blindness: results from west Africa, south India and Chile. Eye (Lond) 7(1): 184-188.
-
Dandona L, Williams JD, Williams BC, Rao GN (1998) Population-based assessment of childhood blindness in southern India. Arch Ophthalmol 116(4): 545-546.
-
Graw J (2004) Congenital hereditary cataracts. Int J Dev Biol 48: 1031-1044.
-
Reddy MA, Francis PJ, Berry V, Bhattacharya SS, Moore AT (2004) Molecular genetic basis of inherited cataract and associated phenotypes. Surv Ophthalmol 49(3): 300-315.
-
Shiels A, Bennett TM, Hejtmancik JF (2010) Cat-Map: putting cataract on the map. Mol Vis 16: 2007-2015.
-
Shiels A, Hejtmancik JF (2013) Genetics of human cataract. Clin Genet 84(2): 120-127.
-
Santhiya ST, Kumar GS, Sudhakar P, Gupta N, Klopp N et al. (2010) Molecular analysis of cataract families in India: new mutations in the CRYBB2 and GJA3 genes and rare polymorphisms. Mol Vis 16: 1837-1847.
-
Kumar GS, Kyle JW, Minogue PJ, Kumar KD, Vasantha K, et al. (2012) An MIP/AQP0 mutation with impaired trafficking and function underlies an autosomal dominant congenital lamellar cataract. Exp Eye Res 110:136-41.
-
Minogue PJ, Liu X, Ebihara L, Beyer EC, Berthoud VM (2005) An aberrant sequence in a connexin46 mutant underlies congenital cataracts. J Biol Chem 280(49): 40788-40795.
-
Berthoud VM, Minogue PJ, Guo J, Williamson EK, Xu X, et al. (2003) Loss of function and impaired degradation of a cataract-associated mutant connexin50. Eur J Cell Biol 82(5): 209-221.
-
Minogue PJ, Beyer EC, Berthoud VM (2013): A connexin50 mutant, CX50fs, that causes cataracts is unstable, but is rescued by a proteasomal inhibitor. J Biol Chem 288(28): 20427-20434.
-
Beyer EC, Ebihara L, Berthoud VM (2013) Connexin mutants and cataracts. Front Pharmacol 4: 43.
-
Mathias RT, White TW, Gong X (2010) Lens gap junctions in growth, differentiation, and homeostasis. Physiol Rev 90(1): 179-206.
-
Hansen L, Mikkelsen A, Nurnberg P, Nurnberg G, Anjum I, et al. (2009) Comprehensive mutational screening in a cohort of Danish families with hereditary congenital cataract. Invest Ophthalmol Vis Sci 50(7): 3291-3303.
-
Arora A, Minogue PJ, Liu X, Reddy MA, Ainsworth JR, et al. (2006) A novel GJA8 mutation is associated with autosomal dominant lamellar pulverulent cataract: further evidence for gap junction dysfunction in human cataract. J Med Genet 43(1): e2.
-
Vanita V, Singh JR, Singh D, Varon R, Sperling K (2008) A mutation in GJA8 (p.P88Q) is associated with "balloon-like" cataract with Y-sutural opacities in a family of Indian origin. Mol Vis 14: 1171-1175.
-
Gong X, Agopian K, Kumar NM, Gilula NB (1999) Genetic factors influence cataract formation in α3 connexin knockout mice. Dev Genet 24(1-2): 27-32.
-
Gerido DA, Sellitto C, Li L, White TW (2003) Genetic background influences cataractogenesis, but not lens growth deficiency, in Cx50-knockout mice. Invest Ophthalmol Vis Sci 44(6): 2669-2674.
-
Graw J, Schmidt W, Minogue PJ, Rodriguez J, Tong JJ, et al. (2009) The GJA8 allele encoding CX50I247M is a rare polymorphism, not a cataract-causing mutation. Mol Vis 15: 1881-1885.
-
Xu X, Berthoud VM, Beyer EC, Ebihara L (2002) Functional role of the carboxyl terminal domain of human connexin 50 in gap junctional channels. J Membr Biol 186(2): 101-112.
-
De Rosa AM, Mui R, Srinivas M, White TW (2006) Functional characterization of a naturally occurring Cx50 truncation. Invest Ophthalmol Vis Sci 47(10): 4474-4481.
-
Wang Z, Schey KL (2009) Phosphorylation and truncation sites of bovine lens connexin 46 and connexin 50. Exp Eye Res 89(6): 898-904.
- Screening of Hospital Staff During World Glaucoma Week in a Tertiary Eye Care Centre
- Angioid Streaks with Macular Neovascularization: Clinical Insights from Two Cases
- Giant Kissing Naevus: An Oculoplastic Challenge
- Why Freedom of Vision Should Not Cost the Freedom of Feeling - LASIK in the Climate of Change
- Asymmetric Optic Nerve with Small Disc and Large Cup: A Rare and Challenging Case of Unilateral Optic Nerve Hypoplasia
- Large Angle Exotropia in a Child: A Case Report