Inflammatory Myofibroblastic Tumor of Lacrimal Gland Rare Tumor in Rare Location in Different Age Groups
Inflammatory myofibroblastic tumor IMT is a rare mesenchymal neoplasm graded as intermediate biological potential. The head and neck region constitute only 5% of IMT and orbit is the rarest anatomical site of this region. In literature, most cases are reported from abdomen followed by lungs. No age is immune for IMT but has special predilection for children and young adults without any sex preference. The present article reports two cases and one case was a 2 years old baby girl who presented with complete ptosis with proptosis of right eye. Another patient was a 50-years old lady who presented with fullness of left upper eye lid along with mild ptosis. In both cases lacrimal gland was the location of tumor. Patients were well managed surgically and tumor did not recur in 3 years of their follow up. Though it is a rare tumor of orbit, it should be a differential in any inflammatory mass lesion. Surgically well managed cases reduce the recurrence rate and ensure eye salvage
Roy SR¹*, Biswas SK² and Anjum R³
¹Consultant and Head, Orbit, Oculoplasty and Ocular Oncology department, Chittagong Eye Infirmary, Bangladesh ²Consultant and Head, Cornea department, Chittagong Eye Infirmary, Bangladesh ³Associate Consultant, Histopathology and Cytopathology Department, Apollo Imperial Hospitals, Bangladesh
Abbreviations
WHO: World Health Organization; IPT: Inflammatory Pseudotumor; AJCC: American Joint Committee on Cancer; ESR: Erythrocyte Sedimentation Rate; SMA: Smooth Muscle Actin; MSA: Muscle Specific Actin; EMA: Epithelial Membrane Antigen; ALK1: Anaplastic Lymphoma Kinase 1; NSAIDs: Non-Steroidal Anti-Inflammatory.
Introduction
IMT that is Inflammatory myofibroblastic tumor described as one of the rare sarcomas which can develop from different anatomical site [1]. In 2013, World Health Organization (WHO), has classified this tumor as tumor of intermediate grade with a low potential for recurrence but rare potential for metastasis [2] This type of tumor was first identified in 1903 from orbit and named in 1905 as inflammatory pseudotumor (IPT) [3]. The same type of lesion was described from lungs in 1937 by Brunn [4]. This lesion was first named as myofibroblastic tumor (IMT) by Umiker et al. because clinically, radiologically and histopathologically it was mimicking as malignancy as per description of Brunn. Subsequently, the lung IMTs were defined as a separate entity from the diverse inflammatory pseudotumour (IPT) group in 1990 and neoplastic activity of this tumor was detected in 1999 [5, 6]. This tumor has different synonyms such as pseudosarcomatous myofibroblastic proliferation, plasma cell pseudotumour, inflammatory myofibrohistiocytic proliferation, myxoid hamartoma, plasma-cell granuloma etc [1, 7]. Though lung and abdomen are the commonly affected sites, the commonest site for primary lesion is the abdomen with a predilection for mesentery, retroperitoneal area and omentum. Beside this, occurrence of lesion is reported at various anatomical sites. Both the college of American pathologists and the American Joint Committee on Cancer recommended that IMT should be classified according to pTNM of 8th AJCC classification (pathological soft tissue stage classification) and according to this classification the location for primary tumor are grouped as head and neck, trunk and extremities, abdomen and thoracic visceral organs, retroperitoneum and orbit [8]. There are some reports of rare location such as esophagus, pericardium, heart, spinal meninges, adrenal gland, spermatic cord and central nervous system [9]. The world prevalence of IMT ranges from 0.04% to 0.7% [4]. This tumor can occur at any age but most commonly occur in children and young adult with a median age of diagnosis is less than 10 years [10]. Here we described two cases of orbital IMT in different age groups.
Case 1
A 2 years old girl presented with rapid development of proptosis with ptosis of right eye without any history of fever or trauma. According to her parent, the symptom was for 3 weeks and she was almost playful. Examination revealed complete ptosis of right eye with moderate proptosis and downward displacement of the globe (Figure 1A). Right eye lid was inflamed (red and hot) also. Her left eye was normal and systemic examination detected no abnormalities. CT scan of orbit showed a globular lesion involving lacrimal gland area, extending to posterior part of orbit and occupying superolateral and superior orbit, and both muscles (lateral rectus and superior rectus) cannot be separated from the lesion (Figure 2A). The lesion became mildly hyperdense after taking contrast. Her complete blood count showed some relative lymphocytosis and peripheral blood film was normal. The lesion was removed completely by lateral orbitotomy and size of the lesion about 3.9 x 2.1x1.6 cm (Figure 3A). Histopathologically, the lesion was composed of spindle or stellate cells arranged in fascicles in a fibro-collagenous stroma. Spindle cell’s cytoplasm was stained pale pink in Hematoxylin and Eosin stain with elongated nucleus. There was myxoid area in the stroma and was infiltrated with variable proportion of lymphocytes, plasma cells and histiocytes. Dilated vascular channels were observed with perivascular infiltration of inflammatory cells. There was no mitosis or necrosis and lymphoid aggregation. The microscopic feature was consistent with inflammatory myofibroblastic tumor. Post operatively patient developed complete ptosis (Figure 1B) as a complication of the surgery. The ptosis gradually improved and no recurrence was seen in three years of follow up.
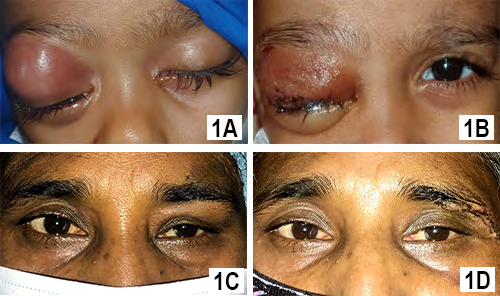
Figure 1A: Preoperative Presentation of 2 Years Old Girl Showing Right Inflamed Eye Lid with Proptosis.
Figure 1B: Immediate Post Operative Image Showing Complete Ptosis with Reduced Inflammation.
Figure 1C: Preoperative Presentation of 50 Years Old Lady with Left Upper Lid Swelling.
Figure 1D: Immediate Post Operative Image Showing Reduced Swelling.
Case 2
A 50 years old female presented with painless swelling of left upper lid for 6 months. She had no systemic illness and no history of trauma. Examination revealed 6/12 vision in both eyes which improved to 6/6 with +0.75D spherical correction. Left eye showed mild ptosis of 2 mm with fullness in lateral part of upper lid (Figure 1C). Ocular motility in left eye was mildly restricted in up and left lateral gaze. Palpation of orbit revealed a non -tender firm, globular mass in supero- lateral orbit. CT scan of orbit showed, a circumscribed lesion measuring about 2.0x1.8x1.2 cm involving the left sided lacrimal gland without any compression to globe. There was no bony change or muscular involvement. The lesion was isodense and became mildly hyperdense with contrast (Figure 2B & 2C). Her other ancillary test like complete blood count, random blood sugar and USG of whole abdomen were normal. She was provisionally diagnosed as lacrimal gland pleomorphic adenoma. Per operatively the lesion found solitary with well capsulated and removed by anterior orbitotomy. Histopathologically, there was spindle/ stellate shaped myofibroblasts and inflammatory cells in fibro collagenous stroma. The spindled to stellate cells were ranged haphazardly in short or interlacing fascicles. Cytoplasm was generally sparse or pale eosinophilic with plump and oval nuclei. In stroma, there were abundance of vessels and inflammatory cells. Lymphocytes, and plasma cells were more prominent cell and neutrophil as well as eosinophils were occasional. (Figure 3B & 3C). Lipid -laden histiocytes was also present. No mitosis and lymphoid aggregation were seen. These histologic features support the diagnosis of inflammatory myofibroblastic tumor. Lid swelling reduced seven days after surgery with some residual ptosis which gradually improved (Figure 1D). The patient showed no recurrence in 4 years follow up.
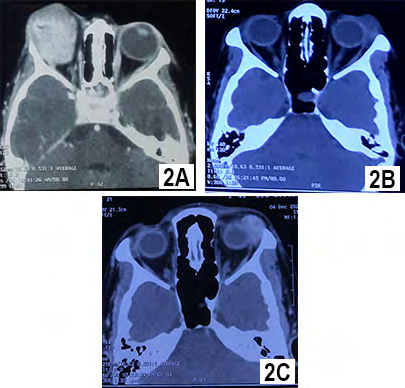
Figure 2A: CT scan of Orbit Of 2 Years Girl, showing a Globular Lesion Occupying Superolateral and Superior Orbit Involving Lacrimal Gland Area and Extending to Posterior Part of Orbit Which Cannot Be Separated from Lateral Rectus and Superior Rectus Muscle. Figure 2B: CT scan of Orbit Of 50 Years Female, showing a Circumscribed Lesion Involving the Left Sided Lacrimal Gland Which is Isodense. Figure 2C: The Lesion Became Mildly Hyperdense with Contrast. The Lesion Did Not Compress the Globe, Not Involved the Muscle and Without Any Bony Change.
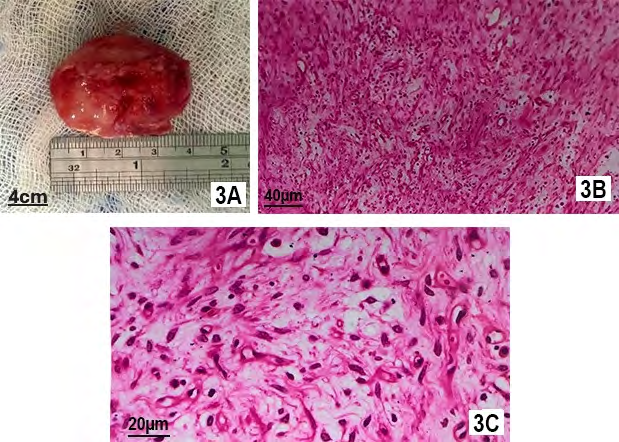
Figure 3A: Excised Lesion from 2 Years Old Girl. The Lesion is About 4 mm X 2.5 mm Size and Whitish in Color. Figure 3B: Microscopic Feature with Hematoxylin and Eosin Stain of 20 Times Magnification Showing Spindle/ Stellate Shaped Myofibroblasts and Inflammatory Cells in Fibro Collagenous Stroma. The Elongated Spindle Cells Were Haphazardly Organized in Short or Interlacing Fascicles. Cytoplasm Was Generally Sparse or Pale Eosinophilic with Plump, Oval Nuclei. In Stroma, There Were Abundance of Vessels and Inflammatory Cells. Lymphocytes, and Plasma Cells Were More Prominent Cell and Neutrophil as Well as Eosinophils Were Occasional. Lipid -Laden Histiocytes was also Present. Figure 3C: Same Microscopic Feature of 40 Times Magnification.
Discussion
Inflammatory myofibroblastic tumor may arise at different sites of head and neck region such as throat, tonsil, parapharyngeal space, thyroid gland, parotid gland, mouth, maxillary sinus, nasal cavity lacrimal gland, orbit and even develop from ocular surface [11, 12, 13]. The incidence of IMT in head and neck region is about 5% of total IMT cases. Most reported cases of IMT in the head and neck region were from orbit, which presented as idiopathic orbital inflammation [12]. Regarding pathogenesis of this tumor, it is thought to be idiopathic but some associations like vasculitis, infection with mycobacteria and thrombosis of inferior vena cava has reported [13]. The definite cause is still not identified and possible causes include trauma, surgery, autoimmune reaction lymphoma (T and B cell type).
The first orbital IMT was reported in 2005, who was a 10-year-old boy [14]. Article search ups showed there are about 55 primary orbital IMT cases has been reported in English language literature up to 2024. These include, 17 single case reports and two case series of 25 cases and 13 cases [15]. These two-case series were retrospective study of 20 and 13 years respectively.
The ocular presentation of IMT has been reported as orbital mass of extraconal or intraconal region, enlargement of extraocular muscle, enlarged lacrimal gland, conjunctival mass and phthisis bulbi. Orbital involvement is usually unilateral but bilateral involvement may also occur [13]. Orbital presentation of IMT may be different and one of largest case series showed the commonest presentation was proptosis (76%). Other presentations were ptosis, diplopia, periocular swelling, pain and redness [16]. Regarding location of tumor, interestingly none of the tumor located at inferior part of orbit in the same series and the commonest location was superolateral orbit (60%) followed by superomedial (24%) and superior (16%). In the current case reports, tumors were located at superolateral orbit mostly due to involvement of lacrimal gland. The differentials of this tumor depend on location of lesion, age and presentation. The common differentials may be orbital cellulitis due to inflammation, rhabdomyosarcoma, lacrimal gland inflammation or tumor and lymphoma.
Literature search ups showed, children and young adults are commonly affected and no predilection for sex and race. In the largest case series of orbital IMT, the age range was 5 years to 76 years and most affected age group was 60-69 years [16]. In the second series, the age range was 2 to 54 years and the most affected group was 25 to 34 years [15].
One recent review stated that, though IMT can develop at any age but most patients presented below 40 years [9]. It was found that children and adolescents are affected more in extrapulmonary IMT, but orbital case series showed most patients were older than 18 years. In literature, the incidence of this tumor stated equal for male and female but both the orbital case series demonstrated a female predilection and our two reported cases were female too [15].
To diagnose this tumor, laboratory examination and imaging provide less clue. Occasionally, some abnormalities such as raised erythrocyte sedimentation rate (ESR), anemia (hypochromic, normocytic), thrombocytosis, hypergammaglobulinemia were reported which became normal immediate after removal of these tumor [9]. Imaging studies are also not so much conclusive. In computed tomography, this lesion appeared as homogenous to heterogenous mass with mild to moderate enhancement with contrast and may be well defined to infiltrative appearance. Enhancement depends on fibrosis of the tumor. Though calcification is generally absent, its presence might think about other malignancy. It may cause bony destruction if located near the bone [9]. In MRI, the lesion is hypointense in T1 and become hyperintense in T2 scan. More enhancement with contrast indicates more aggressiveness of the tumor and peritumoral oedema indicate benign nature which helps in outlining surgical resection [9]. Diagnosis of IMT mainly based on tissue sampling and histopathological examination. Almost all orbital cases are diagnosed by histopathological examination [13].
IMT usually consists of different inflammatory cellular component along with long and slender cells which are looking like spindle. These cells are found in increased number but the differentiation is benign and usually no sign of malignancy or atypia is found. The cells are organized in interlacing pattern in stroma of either myxoid or collagenous type. These spindle cells are mainly resembled myofibroblasts and fewer are fibroblasts which may also display as ganglion-like cells. The predominant inflammatory cells are lymphocytes, plasma cells and occasionally eosinophils, neutrophils which are present in variable proportion [13, 9]. There are three basic histologic patterns of IMT and may found in the same tumor but one of them become predominant depending on distribution of cells and stroma, such as- a) myxoid, b) compact spindle cell and c) fibrous hypocellular. In myxoid pattern, rounded spindle cells are loosely arranged in an edematous or myxoid stroma where vascular component are prominent and plasma cells, neutrophils, and eosinophils are fewer than other patterns [9]. In compact spindle cell pattern, the myofibroblasts are plum to gangliocytic appearance and arranged as storiform fascicles embedded in collagenous, myxoid, loose stroma. The inflammatory cells are abundant. Aggregates of plasma cells, lymphoid follicles and inflammatory infiltrate mixed with spindle cells can also be seen here. In hypocellular pattern, the long, slender (spindle) cells are embedded in a dense stroma of collagen with few cells of inflammation such as plasma cells, lymphocytes and eosinophils. Cell division (mitoses) may present [9]. These three patterns have histopathological similarity of nodular fasciitis, a variety of spindle cell neoplasms and a scar or desmoid fibromatosis respectively [12]. The association of hemorrhage, vascular invasion, necrosis and calcification is usually not found in IMTs but focal calcification of metaplastic or dystrophic variety may found which indicate cellular atypia and recurrence or malignant transformation [10].
To minimize confusion, immunohistochemistry has an important role. Microscopically, the spindle cells of IMT are myofibroblasts comprising feature of both fibroblast and smooth muscle cells. These cells show positive expression of smooth muscle actin (SMA), muscle specific actin (MSA) and Vimentin allowing IMT to be distinguished from other lesions those comprising cells of fibroblast and smooth muscle cell origin and support the diagnosis of IMT [12]. In IMT, spindle cells show expression of 80 to 90% for smooth muscle actin (SMA), 60 to 70% for muscle specific actin (MSA) and staining for Vimentin is typically strong and diffuse [9]. It may show positive or negative staining for desmin, H- caldesmon, CD10, BCL6 transcriptional corepressor but show no reaction for S-100 protein, CD117 and epithelial membrane antigen(EMA) [17].
Recently genetic and molecular biology playing an important role in diagnosis of IMT. About 50% to 70% of IMT show anaplastic lymphoma kinase 1(ALK 1) rearrangements in p21 to p23 region of chromosome 2 [16]. This prevalence is more for lungs tumor followed by gastro-intestine and liver, urinary bladder, intrabdominal site, trunk area, mediastinum and last of all head and neck. The reported orbital cases are mostly ALK negative. For lacrimal gland IMT, IgG4 disease can be a differential. In lung IMT, sometimes it is confused with IgG4 related IPT (Inflammatory pseudotumor) of lung. In eosin and hematoxylin stain, fibrosis, lymphoid aggregates and obliterated vasculitis (phlebities) are prominent features for IgG4 related lung IPT. Plasma cells are mainly IgG4 positive in IPT rather than IMT. Regarding IHC, SMA showed no significant difference between these two tumors but IPT do not show any ALK rearrangement. In such situation, ALK is considered as exclusion for IPT but not as a diagnostic indicator for IMT as IMT can be ALK negative also. FISH (Fluorescein In Situ Hybridization) is recommended for negative ALK of suspected IMT cases. Some researcher stated that, in IMT cases IgG4- positive plasma cells number and ratio of IgG4+/IgG+ plasma cell ratio are generally lower than IgG4-related diseases. Recently there are three criteria are proposed on which the Histopathology – IMT usually consists of spindle cells (mainly myofibroblasts) with various inflammatory cellular component (predominant inflammatory cells are lymphocytes, plasma cells and occasionally eosinophils, neutrophils). The differentiation is benign and usually no sign of malignancy or atypia is found. The association of hemorrhage, vascular invasion, necrosis and calcification is usually not found in IMTs. Lymphoid aggregates and obliterated vasculitis (phlebities) are not prominent in IMT. Presence of a smaller number of IgG4 positive plasma cell.
IHC - IMT shows positive expression of smooth muscle actin (SMA, about 80-90%); muscle specific actin (MSA, about 60-70%) and Vimentin (strong and diffuse) allowing IMT to be distinguished from other lesions those comprising cells of fibroblast and smooth muscle cell origin. No reaction for S-100 protein, CD117 and epithelial membrane antigen (EMA). About 50% to 70% of IMT show anaplastic lymphoma kinase 1(ALK 1) rearrangements. FISH (Fluorescein In Situ Hybridization) is recommended for negative ALK of suspected IMT cases.
diagnosis of these two tumors depend, namely-1. Lymphocyte and plasma cell count, 2. Number of IgG4 positive plasma cell and 3. ALK-1 expression [18].
Beside ALK1, other genetic alterations have been reported and as ALK fusion partners, more than ten genes were found [19]. Among them ROS1 and RET gene are common [9]. Though ALK1 expression is highly specific but it is not 100% sensitive and no established correlation has been identified between ALK positivity and tumor size, mitotic rate, cellularity, necrosis or atypia. But ALK positive IMTs shows less recurrence, less aggressive and may have a favorable outcome [13]. ALK negative IMT is found in older age group and has a higher risk of recurrence and metastasis. Our both cases were diagnosed histopathologically and serum IgG4 antibody was within normal limit. It was our limitation that we did not have the facilities for immuhistochemistry.
The first line of treatment of IMT is total surgical excision or wide local excision. Complete surgical excision of tumor with surrounding healthy tissue is typically curative and 5 years survival rate was reported about 91% by Sagar et al. and Iwai et al [9]. Both the reported case of current article were surgically managed. Radiotherapy or chemotherapy occasionally combined with systemic steroid are applied when resection margin is positive or incompletely resected tumor. IMT seems to be radiosensitive tumor and conventional dose is 45 Gy in different fraction. This tumor even showed response to low dose of radiotherapy than the conventional dose. Regarding chemotherapy, there are conventional chemotherapy and targeted therapy for positive ALK expression. The conventional chemotherapies are like non -small cell sarcomas regimes, predominantly with anthracycline-based chemotherapy. The patients who have advanced IMT, the unresectable tumor or metastatic condition, the systemic therapies are specifically advised [9]. Recently this classic therapy concept has been changed and replaced by combination therapy. Along with anaplastic lymphoma kinase inhibitor, corticosteroids or NSAIDs (non- steroidal anti-inflammatory) combination is the replacement therapy which reduce the risk of recurrence and improve prognosis. The first generation ALK inhibitors is crizotinib, and next to that, second and third generation are proposed for treatment purpose. The second- generation medications are ceritinib, alectinib, ensartinib, and brigatinib, where’s lorlatinib is the third- generation medicine. In case of drug resistance associated with first-generation inhibitors, lorlatinib has shown efficacy to reduce tumor burden [9, 12]. Very recently there was a case report of using immunotherapy in a IMT patient with negative ALK expression and hoping that immunotherapy may prove to be an important therapeutic option in future [9].
Its prognosis depends on anatomical location, resectability and number either solitary or multinodular tumor. After complete surgical excision there are 2% to
25% chances of recurrence based on anatomical location specially less in lungs and more in extrapulmonary region. If resection is incomplete at head and neck region, the chances of recurrence may high as 50% or more [12]. The overall recurrence rate remains low after complete resection and a 10-year survival rate is around 80% [20].
Conclusion
Orbit and lacrimal gland are rare location for inflammatory myofibroblastic tumor. Though it is rare and has predilection for younger age, it may be a differential of any age affected by orbital tumor. Proper diagnosis and skilled surgery can reduce recurrence and save vision and eye.
References
-
Boudhas A, Allaoui M, El Asri F, Rharrassi I, El Ochi MR, et al. (2017) Inflammatory myofibroblastic tumor of the lacrimal gland: case report of an exceptional location. BMC Clin Pathol 17:12.
-
Jo VY, Fletcher CD (2014) WHO classification of soft tissue tumours: an update based on the 2013 (4th) edition. Pathology 46(2): 95-104.
-
Kamili MA, Ali G, Dar IH, Dar SH, Wazir HS, et al. (2009) Orbital pseudotumor. Oman J Ophthalmol 2(2): 96-99.
-
Panagiotopoulos N, Patrini D, Gvinianidze L, Woo WL, Borg E, et al. (2015) Inflammatory myofibroblastic tumour of the lung: a reactive lesion or a true neoplasm?. J Thorac Dis 7(5): 908-911.
-
Pettinato G, Manivel JC, De Rosa N, Dehner LP (1990) Inflammatory myofibroblastic tumor (plasma cell granuloma). Clinicopathologic study of 20 cases with immunohistochemical and ultrastructural observations. Am J Clin Pathol 94(5): 538-546.
-
Griffin CA, Hawkins AL, Dvorak C, Henkle C, Ellingham T, et al. (1999) Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res 59(12): 2776-2780.
-
Sa HS, Ji JY, Suh YL, Kim YD (2005) Inflammatory myofibroblastic tumor of the orbit presenting as a subconjunctival mass. Ophthalmic Plast Reconstr Surg 21(3): 211-215.
-
Vounckx M, Jansen YJL, Fadaei S, Geers C, De Pauw V, et al. (2023) Unraveling the spectrum of inflammatory myofibroblastic tumors in the lung: A comprehensive case series highlighting endobronchial, pleural, and lung parenchymal tumors. JTCVS Open17: 297-305.
-
Chmiel P, Słowikowska A, Banaszek Ł, A Szumera- Ciećkiewicz, B Szostakowski (2024) Inflammatory myofibroblastic tumor from molecular diagnostics to current treatment. Oncol Res 32(7): 1141-1162.
-
Dehner C, Dehner L (2025) Inflammatory myofibroblastic tumor, Soft Tissue, PathologyOutlines.com, USA.
-
Niknejad M (2025) Inflammatory myofibroblastic tumors of the head and neck, Radiopaedia.org, Australia.
-
Zhao J, Han D, Gao M, M Liu, C Feng, et al. (2020) Inflammatory myofibroblastic tumor of the neck with thyroid invasion: a case report and literature review. Gland Surg 9(4): 1042-1047.
-
Shah S, Badhu BP, Lavaju P, Pradhan A (2015) Ocular Inflammatory Myofibroblastic Tumor in the Left Eye with Phthisis Right Eye: A Rare Occurrence in a Child. Case Rep Ophthalmol Med 2015: 281528.
-
Sa HS, Ji JY, Suh YL, Kim YD (20050 Inflammatory myofibroblastic tumor of the orbit presenting as a subconjunctival mass. Ophthalmic Plast Reconstr Surg 21(3): 211-215.
-
Guo S, Wang S, Chen C, X He, B Yang, et al. (2024) Inflammatory Myofibroblastic Tumor of the Orbit: A Case Series and Literature Review. J Inflamm Res 17: 11029-11039.
-
Strianese D, Tranfa F, Finelli M, Iuliano A, Staibano S, et al. (2018) Inflammatory myofibroblastic tumor of the orbit: A clinico-pathological study of 25 cases. Saudi J Ophthalmol 32(1): 33-39.
-
Siemion K, Reszec-Gielazyn J, Kisluk J, Roszkowiak L, Zak J, et al. (2022) What do we know about inflammatory myofibroblastic tumors? - A systematic review. Adv Med Sci 67(1): 129-138.
-
Zhu L, Li J, Liu C, W Ding, F Lin, C, et al. (2017) Pulmonary inflammatory myofibroblastic tumor versus IgG4-related inflammatory pseudotumor: differential diagnosis based on a case series. J Thorac Dis 9(3): 598-609.
-
Gros L, Dei Tos AP, Jones RL, Digklia A (2022) Inflammatory Myofibroblastic Tumour: State of the Art. Cancers (Basel) 14(15): 3662.
-
Lawrence B, Perez-Atayde A, Hibbard MK, BP Rubin, P Dal Cin, et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol 157(2): 377-384.
- Screening of Hospital Staff During World Glaucoma Week in a Tertiary Eye Care Centre
- Angioid Streaks with Macular Neovascularization: Clinical Insights from Two Cases
- Giant Kissing Naevus: An Oculoplastic Challenge
- Why Freedom of Vision Should Not Cost the Freedom of Feeling - LASIK in the Climate of Change
- Asymmetric Optic Nerve with Small Disc and Large Cup: A Rare and Challenging Case of Unilateral Optic Nerve Hypoplasia
- Large Angle Exotropia in a Child: A Case Report