A Curious Cluster of Cholesteatoma: A Family Study
Objective: We report a rare case of family clustering of cholesteatoma. Method: Case reports of three first-degree relatives in a family affected with cholesteatoma and a review of world literature on hereditary causes for cholesteatoma are presented. Results: The family consists of parents and two siblings, of whom the father and both siblings (a daughter and a son) were surgically treated for cholesteatoma. All cholesteatomas in this family cluster were acquired. Conclusion: Family clustering of acquired cholesteatoma is rare. This report indicates the interplay of hereditary factors along with environmental factors in the genesis of cholesteatoma.
Introduction
Cholesteatoma is a self-expanding disease of the middle ear, composed of a stratified keratinizing squamous epithelium with accumulated keratin debris, which has a propensity to erode bone [1].
Cholesteatoma may be congenital or acquired. When cholesteatoma is found behind an intact tympanic membrane, it is called a congenital cholesteatoma [2]. If a congenital cholesteatoma expands laterally and erodes the tympanic membrane, then it becomes difficult to differentiate it from acquired cholesteatoma.
The acquired type of cholesteatoma originates due to an inward growth of squamous epithelium of the tympanic membrane or external auditory canal into the middle ear cavity. In children with a history of otitis media with effusion, a retraction of pars flaccida is developed in 15 to 35%, but cholesteatoma is developed only in 0.1 to 2%. [3]. Although otitis media with effusion is closely associated with cholesteatoma, it is still not fully understood why some patients develop cholesteatoma while the vast majority do not. Here, in the given family cluster of cholesteatoma, three first-degree relatives in a family of four (father, daughter, and son) are affected with the disease and underwent surgical treatment between 2001 and 2023. This generated a question that is there a genetic predisposition to cholesteatoma within families? Or is it the shared environment that predisposed them to this disease? The medical histories of the three family members with cholesteatoma are as described.
Case 1 (Father)
49-year-old male presented with symptoms of right-sided otorrhoea and hearing loss. On examination, he had a right- sided postero-superior retraction pocket filled with keratin flakes, and the fundus of the sac was not seen (Figure 1a). In the left ear, he had a grade IV retraction of Pars Tensa with a shallow posterosuperior retraction pocket whose fundus was seen with no collection of keratin debris. He was diagnosed with a case of bilateral chronic otitis media, squamous type, right active and left inactive. He underwent right intact canal wall mastoidectomy with type III tympanoplasty. Per- operatively, it was seen that the lenticular process and long process of incus were eroded and the stapes superstructure was absent. The Cholesteatoma sac was removed; incus was removed, refashioned, and interposed between the stapes footplate and malleus. Tympanic membrane remnant was reinforced with temporalis fascia graft. Pre-operative/post- operative pure tone (air conduction) average (average of 500 Hz, 1KHz, and 2 KHz) in the right ear was 50/ 36 dB.
Case 2 (Daughter)
21 years old female, a medical student with no known comorbidities, presented with complaints of otorrhoea (left side) since childhood and hearing loss (Lt) for the last 02 years. On examination, there was grade IV attic erosion with keratin debris in the attic. She was diagnosed with a case of chronic otitis media (Lt) – squamous type active. She underwent intact canal wall mastoidectomy + type III tympanoplasty (major columella) + lateral semicircular canal fistula repair+ outer attic wall reconstruction under general anesthesia. Per-operatively, a cholesteatoma sac was seen in the attic extending into the anterior epitympanum, supra-tubal recess, aditus, and antrum. The facial canal was dehiscent from the tympanic segment to the 2nd genu with granulation tissue covering it. The Malleus head was partially eroded, but the handle was preserved. Incus and stapes superstructure were absent; the footplate was preserved and mobile. Attic erosion was present (grade IV); granulation tissue was seen extending into the lateral semicircular canal (LSCC), eroding its dome with no perilymph leak (Figures 1b, 2a & 2b). Intact canal wall mastoidectomy and posterior epitympanotomy were done. Cholesteatoma was removed and granulation on the lateral semicircular canal was removed. Temporalis fascia graft was placed by underlay technique and attic reconstruction was done with tragal cartilage. The LSCC fistula was repaired with temporalis fascia and periosteum. Pre-operative/post-operative hearing pure tone average was 33/25 dB.
Case 3 (Son)
17-year-old male, with no known comorbidities presented with complaints of otorrhoea (right side) of 05 years of duration and otalgia (Rt) with vertigo for the last 2 months. Vertigo was episodic, positional, and rotatory, each episode lasted for 15-20 minutes, relieved by lying still and taking medications. On examination, there was an attic cholesteatoma causing grade III attic erosion and the fistula test was positive. He was diagnosed with a case of Chronic otitis media (Rt)- squamous type, active with profound mixed hearing loss and right lateral semicircular canal fistula. He underwent an intact canal wall mastoidectomy with Type III tympanoplasty (Rt) with lateral semicircular canal fistula repair under general anesthesia. During surgery, there was granulation tissue seen in the middle ear, extending into aditus, antrum, and medially extending to LSCC. The mastoid antrum was contracted with an anteriorly placed sigmoid sinus and an inferiorly placed dura. Malleus was present, the long process of incus was eroded and the stapes superstructure was partly eroded (Figures 1c, 3a & 3b). The LSCC fistula was present without a perilymph leak. The granulation tissue over the LSCC was removed and the fistula was covered with temporalis fascia graft and bone dust. Pre- operative/post-operative hearing pure tone average was 116/85 dB. Temporalis fascia graft was used to support the tympanic membrane remnant. Ossiculoplasty was done using refashioned incus.
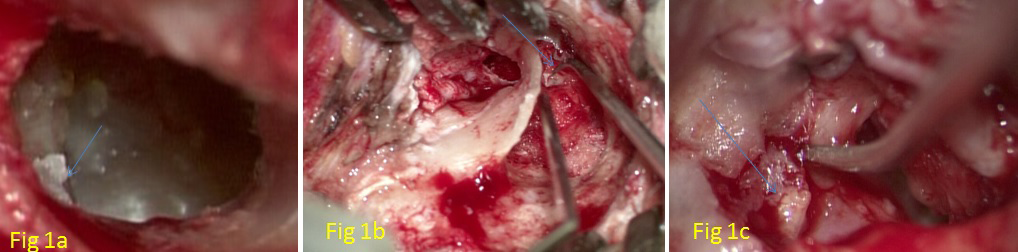
Figure 1a: (Case1- Father) Arrow Pointing to Postero-Superior Retraction Pocket (Right Ear). Figure 1b: (Case 2- Daughter) Arrow Pointing to LSCS Fistula (Left Ear). Figure 1c: (Case 3- Son) Arrow Pointing to Partially Eroded Stapes Superstructure (Right Ear).
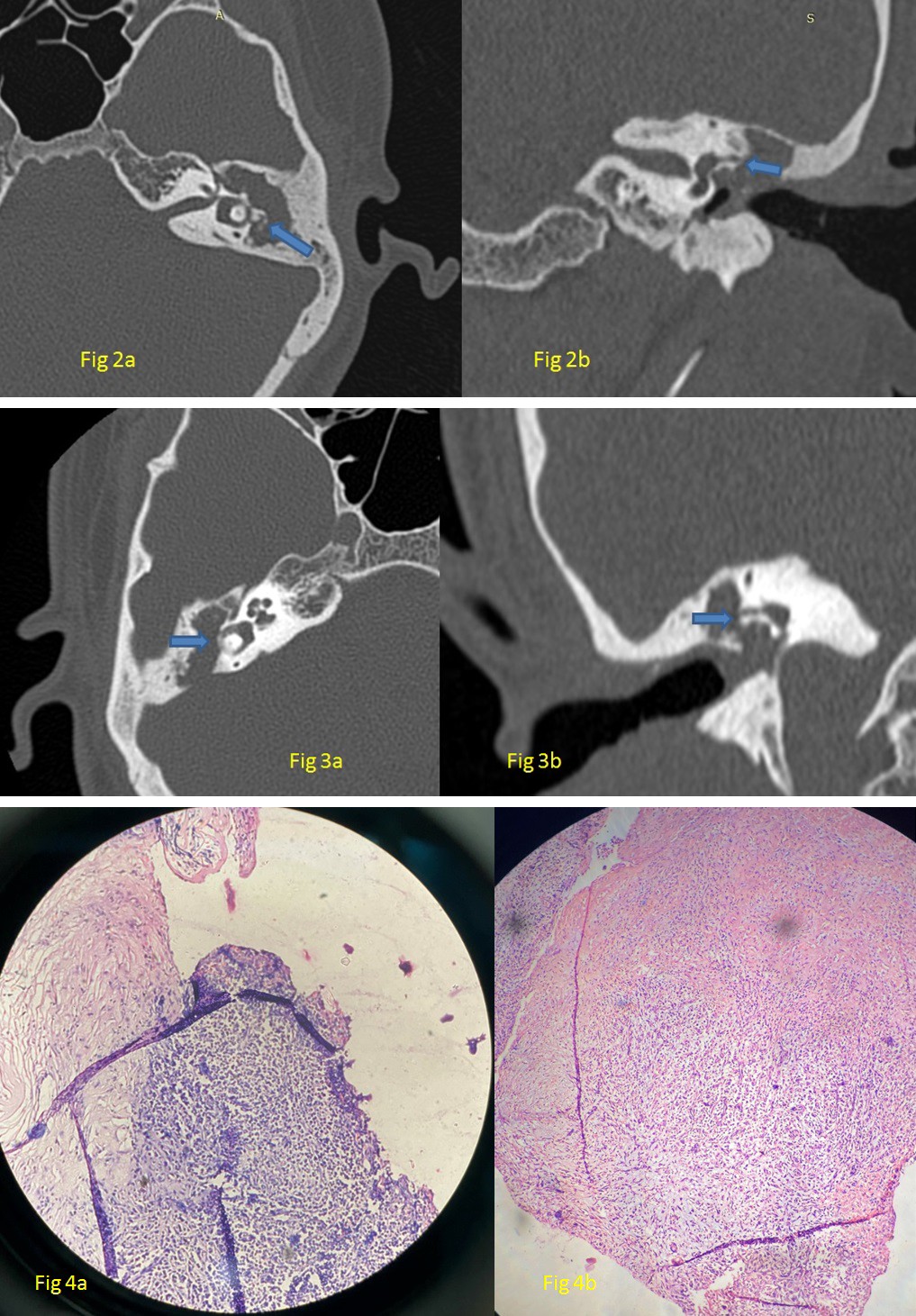
Figure 2a: High Resolution Computed Tomography (HRCT) Temporal Bone of Daughter, Axial Cut Showing Soft Tissue Shadow in LT Attic, Aditus and Antrum with Erosion of LT LSCC Dome (Arrow). Figure 2b: Coronal Section at the Same Level Showing Erosion of Lt Lscc Dome (Arrow).
Figure 3a: HRCT temporal bone of son, axial cut showing soft tissue shadow in RT attic, aditus and antrum with erosion of RT LSCC dome (arrow). Figure 3b: Coronal Section at the Same Level Showing Erosion of RT LSCC Dome (Arrow).
Figure 4a: HPE of Case 2 (Daughter). Figure 4b: HPE of Case 3 (Son), Both Consistent With Cholesteatoma.
The histopathological examination (HPE) of the excised tissue of case 2 (daughter) showed strips of squamous epithelium surrounded by granulation tissue with inflammatory infiltrate comprising of lymphocytes, plasma cells and neutrophils, which was consistent with cholesteatoma (Figure 4a). The HPE report of the excised tissue of case 3 (son) showed keratin flakes and granulation tissue comprising of mixed inflammatory infiltrates in the form of lymphocytes, plasma cells and macrophages, consistent with cholesteatoma (Figure 4b).
Discussion
Cholesteatoma is a rare disorder with an incidence of 5 to 10:100,000 per year in various populations [4, 5, 6]. Therefore, it is difficult to conduct epidemiological studies, hence the causative risk factors are not very well understood. Case reports of familial clustering of cholesteatoma and the association with genetic syndromes point towards the possibility of underlying, but yet not completely identified genetic risk factors [7]. Identifying the genetic factors could augment our understanding of disease biology, and will better aid in screening and therapeutic interventions. Previous studies have indicated that alterations of chromosomes such as aneuploidy of chromosome 8 and trisomy 7 may play important roles in the prognosis of cholesteatoma [8, 9]. Cholesteatoma is reported in patients with branchio- oto-renal syndrome, suggesting the importance of inborn mechanisms [10]. There are case reports giving studies of DNA- based gene sequences that revealed deletion of APC tumour suppressor gene [11] and association of polymorphisms of GJB2 and GJB6 loci which encode for connexions [12]. No conclusions may be safely drawn from such case reports, as they lack a control population and generally the sample size in such reports are small.
Descriptions in a few case report and case series about familial clustering of cholesteatoma are insufficient evidence to explain cholesteatoma as a genetic trait. However, there are few case reports on affected twins [13], families with two or more generations affected [14], and bilateral disease in affected families [15]. In the present family clustering, two among three members had only unilateral disease and two generations were involved with the disease. One of the proposed mechanisms of genetic influence is an altered genetic control of cellular proliferation. Molecular research also suggests that genes might influence epithelial cell behavior in the middle ear. Mesenchymal tissue in the middle ear normally eliminates cellular ingrowths or ectodermal remnants by apoptosis. This process may be defective in cholesteatoma [15]. In a Swedish case-control study (2018) using national registry data, there was a strong association between a family history of cholesteatoma with the risk of a first-degree relative developing cholesteatoma in the middle ear (almost four times higher) [16]. In a recent study by Lee NK et al [17], the salivary DNA of patients with cholesteatoma was subjected to exome sequencing. Tissue samples obtained from these patients during tympanoplasty and mastoidectomy were submitted for mRNA sequencing and differentially expressed genes (DEGs) were analyzed. Rare and probably damaging variants from the generated exome sequence were selected within previously identified DEGs. The candidate genes within these variants were used for network analysis. Such network analysis identified ten cellular pathways which are significant in viral process, protein transport, regulation of cell cycle and catalytic activity, apoptosis, and rhythmic processes. They have hypothesized that these identified genes may be part of important signaling pathways in mucosal response to infections of the middle ear. These may play a role in the onset of cholesteatoma [17].
Some congenital syndromes such as Turner Syndrome, Down Syndrome, and cleft palate pose the affected individuals with an increased risk of otitis media with effusion, which itself is a causative factor for the development of cholesteatoma. Hence, if these syndromes in themselves are associated with an increased risk of cholesteatoma formation is difficult to comment [7]. In this present case series of family cluster, we cannot claim genetic disposition as the only factor for the development of cholesteatoma. There is a possibility of several other independent factors in the family and surrounding environment which might have predisposed them to recurrent upper respiratory tract infection and resultant clinical/ subclinical otitis media with effusion. Anatomical factors such as Eustachian tube anatomy, rhino pharyngeal dimensions, cranial angles, temporal bone pneumatization, and middle ear anatomy have a bearing on the predisposition for the development of cholesteatoma [14]. These factors may have a similarity among family members.
Conclusion
On analyzing a few of the available case reports and case series of familial clustering, cholesteatoma has been shown to run within families in a pattern of oligogenic or monogenic disorder with incomplete penetrance. The probability of development of cholesteatoma will depend on a combination of environmental factors and genetic factors of variable penetrance. However, this adds to the possible etiopathogenesis of the development of cholesteatoma.
Strengths of the Study
Data about the pre-operative, intraoperative, and postoperative findings of all members affected were available and two generations in the same family cluster were studied.
Limitations of the Study
In the research hierarchy, case series provide low-level evidence and the findings of the report are not generalizable. Genetic analysis was not done in this study, as the members of the family were not willing for the same.
References
-
Bhutta MF, Williamson IG, Sudhoff HH (2011) Cholesteatoma. BMJ 342: d1088.
-
Kazahaya K, Potsic WP (2004) Congenital cholesteatoma. Current Opinion in Otolaryngology & Head And Neck Surgery 12(5): 398-403.
-
Schilder AG, Zielhuis GA, Haggard MP, Den Broek PV (1995) Long-term effects of otitis media with effusion: otomicroscopic findings. Am J Otol 16(3): 365-372.
-
Homøe P, Bretlau P (1994) Cholesteatomas in Greenlandic Inuit. A retrospective study and follow-up of treated cases from 1976-91. Arct Med Res 53(2): 86-90.
-
Tos M (1998) Incidence, etiology and pathogenesis of cholesteatoma in children. Adv OtolrhinolLaryngol 40: 110-117.
-
Kemppainen HO, Puhakka HJ, Laippala PJ, Sipila MM, Manninen MP, et al. (1999) Epidemiology and aetiology of middle ear cholesteatoma. Acta Otolaryngol 119(5): 568-572.
-
Jennings BA, Prinsley P, Philpott C, Willis G, Bhutta MF (2018) The genetics of cholesteatoma. A systematic review using narrative synthesis. Clin Otolaryngol 43(1): 55-67.
-
Lavezzi A, Mantovani M, Cazzulo A, Turconi P, Matturri L (1998) Significance of trisomy 7 related to PCNA index in cholesteatoma. Am J Otolaryngol 19(2): 109-112.
-
Yildirim MS, Ozturk K, Acar H, Arbag H, Ulku CH (2003) Chromosome 8 aneuploidy in acquired cholesteatoma. Acta Otolaryngol 123(3): 372-376.
-
Graham GE, Allanson JE (1999) Congenital cholesteatoma and malformations of the facial nerve: rare manifestations of the BOR syndrome. Am J Med Genet 86(1): 20-26.
-
Shaoul R, Rapsin B, Cutz E, Durie P (1999) Congenital cholesteatoma in a child carrying a gene mutation for adenomatous polyposis coli. Journal of pediatric gastroenterology and nutrition 28(1): 100-103.
-
James AL, Chadha NK, Papsin BC, Stockley TL (2010) Pediatric cholesteatoma and variants in the gene encoding connexion 26. The Laryngoscope 120(1): 183- 187.
-
Al Balushi T, Naik JZ, Al Khabori M (2013) Congenital cholesteatoma in identical twins. J Laryngol Otol 127(1): 67-69.
-
Homøe P, Rosborg J (2007) Family cluster of cholesteatoma. J Laryngol Otol 121(1): 65-67.
-
Prinsley P (2009) Familial cholesteatoma in East Anglia, UK. J Laryngol Otol 123(3): 294-297.
-
Bonnard Å, Berglin EC, Wincent J, Eriksson PO, Westman E, et al. (2023) The Risk of Cholesteatoma in Individuals With First-degree Relatives Surgically Treated for the Disease. JAMA Otolaryngol Head Neck Surg 149(5): 390- 396.
-
Lee NK, Cass SP, Gubbels SP, Gomez HZ, Scholes MA, et al. (2023) Novel candidate genes for cholesteatoma in chronic otitis media. Front Genet 13:1033965.
- 4th Branchial Cleft Sinus Anomaly Presenting as Recurrent Thyroid Abscess in A Child: A Case Report
- Parotid Duct Injury Repaired Using an Angiocatheter Stent: A Case Report
- Organization and Functionality of the Referral and Counter-Referral System for ENT Disorders in District Hospitals of N’Djamena, Chad: A Cross-Sectional Analytical Study
- Facial Metastases from a Gastrointestinal Stromal Tumor: A Case Report
- Panorama of Ent Cancers and Literature Review: Epidemiological Profile and Therapeutic Management
- Could Antimicrobial Resistance Prove to Be Both a Threat and an Opportunity for us?