USP30 Inhibitors and Mitophagy, a Cellular Power Couple against Metabolic-Associated Fatty Liver Disease
Mitophagy is a selective process by which damaged or dysfunctional mitochondria are specifically targeted for degradation and removal by the cell. It prevents the accumulation of dysfunctional mitochondria, which can otherwise contribute to cellular stress and diseases such as neurodegenerative disorders and certain cancers. Ubiquitination marks proteins for degradation by the proteasome or lysosomes of the autophagy machinery. Ubiquitin-Specific Peptidase 30 (USP30) on the other hand, has been identified as a negative regulator of mitophagy. It counteracts the process of ubiquitination by removing the ubiquitin tags from proteins on the mitochondrial surface and prevents the degradation of damaged or dysfunctional mitochondria leading to cellular stress. Inhibiting USP30 activity has been shown to promote mitophagy and a potential approach to managing certain neurodegenerative diseases. Although impaired mitophagy and mitochondrial dysfunction have been linked to the pathogenesis of metabolic-associated fatty liver disease (MAFLD), research into the involvement of USP30 in the pathophysiology of MAFLD or metabolic disorders is still in its early stages. As a result, we sought to thoroughly assess the literature to determine the involvement of USP30 in the pathophysiology of MAFLD and whether modulating USP30 activity could potentially be a therapeutic strategy to manage MAFLD.
Abbreviations
MAFLD: Metabolic-Associated Fatty Liver Disease; USP30: Ubiquitin-Specific Peptidase 30; MASH: Metabolic Dysfunction-Associated Steatohepatitis; FDA: Food and Drug Administartion; OMM: Outer Mitochondrial Membrane; TOM 20: Translocase of Outer Membrane 20; PINK1: PTEN- Induced Putative Kinase 1; DUB: Deubiquitinase Enzyme; USP30: Ubiquitin-Specific Protease 30; MTX652; HFD: High- Fat Diet; HCC: Hepatocellular Carcinoma; RCR: Respiratory Control Ratio; ACLY: ATP Citrate Lyase; and FASN: Fatty Acid Synthase; LKO: Liver-Specific USP30 Knockout.
Introduction
Metabolic-associated fatty liver disease (MAFLD) formerly known as non-alcoholic steatohepatitis (NAFLD) is a global epidemic impacting more than 30% of the world’s population. A more severe form of disease is called metabolic dysfunction-associated steatohepatitis (MASH). It develops when the accumulation of fat and inflammation in the liver leads to scarring and possibly liver failure. In March 2024, the FDA gave fast-track approval to Madrigal’s Resmetirom (Rezdiffra), making it the first-ever licensed drug for the management of MAFLD. However, even with an approved drug available for prescription, the path ahead for these patients remains challenging because, in phase III testing, Resmetirom improved key readouts of liver pathology in just 25–30% of patients [1]. Therefore, there is a continuous need for better and better drugs.
The recent understanding of MAFLD has progressed from the two-hit theory to the multiple- hit theory, including impaired lipid metabolism, oxidative damage, genetic factors, and components of the gut-liver axis [2]. In addition, mitochondrial dysfunction has also been found to play an important role in the pathogenesis of MAFLD. Mitochondrial dysfunction is characterized by structural abnormalities, oxidative stress, and disruption of the mitochondrial respiratory chain and has been reported to intensify the pathogenesis of MAFLD [3].
Imbalance of PINK1/Parkin signaling by USP30 leads to Impaired Mitophagy
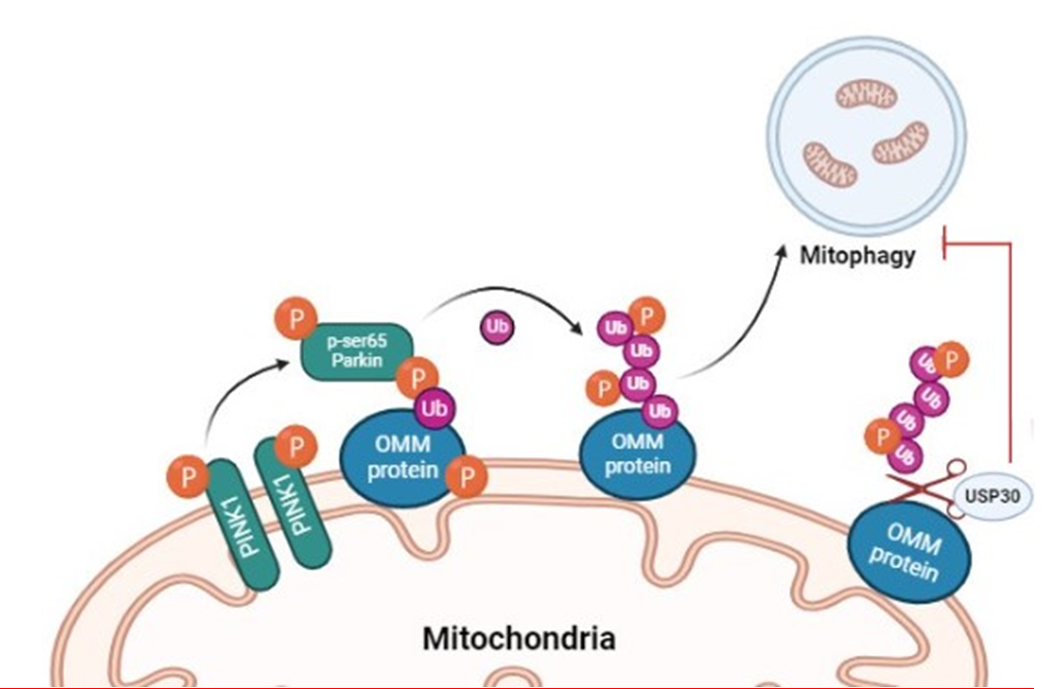
One key factor contributing to mitochondrial dysfunction is the impaired process of mitophagy which is also known as mitochondrial autophagy. This is a quality control mechanism mediated by the ubiquitination of outer mitochondrial membrane (OMM) proteins specifically translocase of outer membrane (TOM20) complex, which is solely responsible for removing damaged mitochondria to maintain cellular mitochondrial quantity and energy homeostasis [4]. Recent reports have confirmed the crucial role of mitophagy in clearing damaged mitochondria in MAFLD patients. A study in mice has revealed that deficiencies in Parkin, an E3 ubiquitin ligase or PTEN- induced putative kinase 1 (PINK1) lead to a loss of mitophagy, worsening the mitochondrial dysfunction and hence accelerating the progression of MAFLD [5]. Another study has confirmed that loss of mitophagy is an early feature of MAFLD and that loss of PARKIN hastens the onset of steatosis, inflammation, and fibrosis [6]. Mutations in Parkin, have also been reported to cause juvenile-onset Parkinsonism probably through the impairment of mitophagy.
A deubiquitinase (DUB) enzyme; Ubiquitin-specific protease 30 (USP30) dampens PINK1/Parkin-dependent amplification of mitochondrial ubiquitination, leading to suppression of mitophagy as shown in Figure 1 [7]. USP30 has been found to play an important role in several cellular events such as pexophagy, BAX/BAK-dependent apoptosis, and IKKβ–USP30–ACLY-regulated lipogenesis/tumorigenesis [8, 9]. According to a recent report, USP30 was also found to accelerate myocardial cell senescence through antagonism of Parkin [10]. While overexpression of USP30 has been documented to negatively influence mitophagy, inhibition of the USP30 has been found to enhance mitophagy [7].
A few small molecule inhibitors of USP30 are currently under investigation for their possible activity in the case of neurological disorders. Out of the ones which have shown significant promise in their pre-clinical and clinical data; MTX652 and MTX325 by Mission Therapeutics are currently under phase I and phase II clinical evaluation for Parkinson’s disease and Acute kidney injury respectively.
Role of USP30 in metabolic-associated fatty liver disease
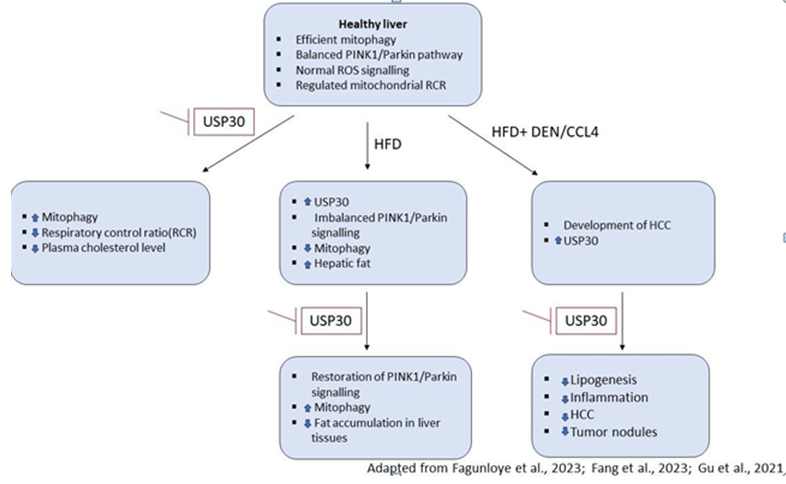
Although mitochondrial dysfunction has been reported to play an important role in the pathogenesis of MAFLD, detailed information about the role of USP30 in the pathophysiology of MAFLD or metabolic disorders is still in its infancy. We, therefore, aimed to carry out a comprehensive review of the literature to find out the role of USP30 in the pathogenesis of MAFLD (Table 1 & Figure 2). The following three lines of evidence may be helpful to build our understanding in this regard.
| Evidence | Effect on MAFLD/HCC | References | |
|---|---|---|---|
| Reduces fat accumulation | USP30 knockout in mice delays liver fat accumulation. | May help manage MAFLD. | [11] |
| Promotes tumorigenesis and lipogenesis | Increased USP30 in high-fat diet mice with HCC. | USP30 knockout reduces tumors, inflammation, and fat production. | [9] |
| Regulates mitophagy and mitochondrial function | Liver-specific USP30 knockout mice have impaired mitochondrial function in healthy mice. | Reduces plasma cholesterol up to 25%. | [12] |
Table 1: Role of USP30 in hepatic fat accumulation.
USP30 knockout mice demonstrate reduced levels of hepatic fat with ageing
In a recent study reported by Fang TSZ, et al. [11] USP30 knockout mice were found to have significantly reduced levels of fat in the liver tissues of one-year-old C57BL/6N mice [13] compared to wild-type mice. This preliminary data suggests the scope of USP30 inhibition in ameliorating the key hallmarks of MAFLD which can be validated further in a detailed study.
IKKβ-USP30-ACLY Axis regulates lipogenesis and tumorigenesis
According to another study by Gu L, et al. [9], mice treated with DEN/CCL4 and maintained on a high-fat diet (HFD) developed hepatocellular carcinoma (HCC) wherein the expression levels of USP30 were found to be significantly high at both mRNA and protein levels. In this study, IκB kinase (IKKβ) was found to phosphorylate and stabilize USP30, which enabled USP30 to deubiquitinate the key lipogenesis-related enzyme; ATP citrate lyase (ACLY) and fatty acid synthase (FASN). Deletion of USP30 in this model showed significant inhibition of lipogenesis, inflammation, and hepatocellular carcinoma. These mice also displayed fewer tumor nodules and reduced tumor burden suggesting the crucial role of IKKβ–USP30–ACLY axis in lipogenesis in addition to HCC.
Regulation of hepatic mitophagy and mitochondrial function by USP30
To investigate the role of USP30-mediated mitophagy in hepatocytes, Fagunloye OG, et al. [12] employed a Cre- loxP approach to carry out liver-specific USP30 knockout (LKO) in C57/BL6 male mice fed on the regular chow diet. Inhibition of USP30 prevents de-ubiquitination of parkin and OMM proteins thereby enhancing the PINK1/parkin- mediated mitophagy which was also reflected in this study. A significant reduction in ADP-stimulated respiration followed by reduced respiratory control ratio (RCR) and an increase in mitochondrial leak respiration was observed in LKO mice. Although no significant changes in liver or plasma triglyceride levels were found, a 25% reduction in plasma cholesterol level was observed.
Conclusion
Inhibition of USP30 emerges as a promising therapeutic target for MAFLD. The available information reported in the literature collectively suggests that: i) USP30 plays a significant role in mitochondrial dysfunction in the liver and ii) inhibition of USP30 can be a potential therapeutic approach to reduce the accumulation of fat in the liver via stabilizing the PINK1/Parkin-mediated mitophagy. Moreover, the IKKβ-USP30-ACLY axis has been implicated in regulating lipogenesis and tumorigenesis, further emphasizing the connection between USP30 and metabolic disorders.
While studies have shown that USP30 inhibition can enhance mitophagy and improve mitochondrial function, more comprehensive investigations are required to elucidate the full spectrum of USP30’s involvement in MAFLD. For future perspective, we further need to study the role of USP30 in mitophagy in greater depth and to investigate whether a small molecular approach of USP30 inhibition can be helpful to manage the complications of MAFLD along with evaluation of USP30 inhibitors in preclinical MAFLD models to assess efficacy and safety and investigation of the combined effects of USP30 inhibition with other therapeutic approaches.
Given the growing prevalence of MAFLD and the limitations of current treatment options, the exploration of USP30 as a therapeutic target presents a promising avenue for addressing this complex metabolic disorder. By advancing our understanding of USP30 and its role in MAFLD, we can develop innovative strategies to improve patient outcomes and mitigate the global burden of this disease.
Conflict of Interests
None
References
-
Harrison SA, Bedossa P, Guy CD, Schattenberg JM, Loomba R, et al. (2024) A Phase 3, randomized, controlled trial of resmetirom in NASH with liver fibrosis. The New England Journal of Medicine 390(6): 497-509.
-
Buzzetti E, Pinzani M, Tsochatzis EA (2016) The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism: Clinical and Experimental 65(8): 1038-1048.
-
Ipsen DH, Lykkesfeldt J, Tveden-Nyborg P (2018) Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cellular and Molecular Life Sciences 75(18): 3313-3327.
-
Luo Z, Yan S, Chao Y, Shen M (2024) Unveiling the mitophagy puzzle in non-alcoholic fatty liver disease (NAFLD): six hub genes for early diagnosis and immune modulatory roles. Heliyon 10(7): e28935.
-
Edmunds LR, Xie B, Mills AM, Huckestein BR, Undamatla R, et al. (2020) Liver-specific Prkn knockout mice are more susceptible to diet-induced hepatic steatosis and insulin resistance. Molecular Metabolism 41: 101051.
-
Undamatla R, Fagunloye OG, Chen J, Edmunds LR, Murali A, et al. (2023) Reduced mitophagy is an early feature of NAFLD and liver-specific PARKIN knockout hastens the onset of steatosis, inflammation and fibrosis. Scientific Reports 13(1): 7575.
-
Rusilowicz-Jones EV, Jardine J, Kallinos A, Pinto- Fernandez A, Guenther F, et al. (2020) USP30 sets a trigger threshold for PINK1–PARKIN amplification of mitochondrial ubiquitylation. Life Science Alliance 3(8): 1-14.
-
Wang F, Gao Y, Zhou L, Chen J, Xie Z, et al. (2022) USP30: Structure, Emerging Physiological Role, and Target Inhibition. Frontiers in Pharmacology 13: 851654.
-
Gu L, Zhu Y, Lin X, Lu B, Zhou X, et al. (2021) The IKKβ-USP30-ACLY Axis Controls Lipogenesis and Tumorigenesis. Hepatology 73(1): 160-174.
-
Pan W, Wang Y, Bai X, Yin Y, Dai L, et al. (2021) Deubiquitinating enzyme USP30 negatively regulates mitophagy and accelerates myocardial cell senescence through antagonism of Parkin. Cell Death Discovery 7(1): 187.
-
Fang TSZ, Sun Y, Pearce AC, Eleuteri S, Kemp M, et al. (2023) Knockout or inhibition of USP30 protects dopaminergic neurons in a Parkinson’s disease mouse model. Nature Communications 14(1): 7295.
-
Fagunloye OG, Bello F, Vandevender A, Sipula IJ, Jurczak MJ (2023) 1572P: Usp30 regulates hepatic mitophagy and mitochondrial function. Diabetes 72(Supplement_1): 1572-P.
-
Choi KM, Jung J, Cho YM, Kim K, Kim MG, et al. (2017) Genetic and phenotypic characterization of the novel mouse substrain C57BL/6N Korl with increased body weight. Scientific Reports 7(1): 14217.
- Gallic and Citric Acid Present in the Peels of Tropical Fruits as an Alternative in the Fight against Cancer
- Treating the Forehead Lines with Combination of Forehead and Glabellar Botulinum Toxin Among Japanese Patients
- Clinical Evaluation of Patients Suffering from Breast Cancer & Determination of Treatment Therapies and Better Strategies Related to Breast Cancer
- Medieval Recipes by Al-Zahrāwī for Heart Palpitations Treatment
- Etiology and Prescription Errors of Myocardial Infarction in Different Health Care Systems of Azad Kashmir
- Early Diagnosis and Multidisciplinary Management of Turner Syndrome: A Paediatric Case Study