Review on Regulator’s Perspective of TGA about Pharmacovigilance during Premarketing and Post Marketing of Medicinal Product
The Review is about Therapeutic Goods Administration (TGA) which is a regulatory authority responsible for ensuring the safety, quality, and efficacy of therapeutic goods in Australia and how it works during the premarketing and post marketing of medicinal products. It clearly explains about the responsibilities of TGA in evaluation of drug safety data during pre-marketing and post-marketing phases, the implementation of risk management plans, and the facilitation of effective communication channels for adverse event reporting.
Abbreviations
TGA: Therapeutic Goods Administration; OTC: Over- The-Counter; USFDA: United States of Food and Drug Administration; EMA: European Medicines Agency; WHO: World Health Organization; IT: Information Technology; ARTG: Australian Register of Therapeutic Goods, PV: Pharmacovigilance; RMP: Risk Management Plan; NCE: New Chemical Entity; DAEN: Database of Adverse Event Notifications; PASS: Post-Authorization Safety Studies; PSURs: Periodic Safety Update Reports.
Introduction
The Therapeutic Goods Administration (TGA) is the medicine and therapeutic regulatory agency of the Australian Government. TGA is a part of Department of Health and Aged Care along common wealth department of health. TGA was established in 1990. Any items that claim to have a therapeutic effect, are involved in the administration of medication, or are otherwise covered by the Therapeutic Goods Act 1989, the Therapeutic Goods Regulations 1990, or a ministerial order, must be approved by the TGA and registered in the Australian Register of Therapeutic Goods. Operating under the Department of Health, the TGA’s mandate encompasses a wide range of products, including prescription medicines, over-the-counter (OTC) medications, vaccines, medical devices, blood products, and more. The TGA plays a crucial role in protecting public health by overseeing the regulation of these therapeutic goods throughout their lifecycle [1].
The TGA collaborates with international regulatory agencies, such as the U.S. FDA, the European Medicines Agency (EMA), and the World Health Organization (WHO).
These collaborations help harmonize regulatory standards, share safety data, and respond to global health threats. Regulators, such as the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), and other national and international bodies, play a pivotal role in pharmacovigilance. TGA was expertise in clinical and scientific decision making [2].
**Plan of TGA**
**Vision and Mission**
Vision: To be a trusted regulator of therapeutic goods, ensuring they are safe, effective, and of high quality, thus protecting and enhancing the health of the Australian community.
Mission: To safeguard and enhance the health of the Australian public through effective and efficient regulation of therapeutic goods [3].
**Strategic Goals**
- Enhance Regulatory Frameworks
- Continuously update and improve regulatory frameworks to keep pace with scientific advancements and emerging technologies.
- Align with international standards and best practices to facilitate global harmonization and streamline regulatory processes.
- Ensure Product Safety and Quality
- Strengthen pre-market evaluation processes to ensure only safe, effective, and high-quality products enter the market.
- Enhance post-market surveillance and risk management strategies to promptly identify and address safety issues.
- Promote Compliance and Enforcement
- Implement robust compliance and enforcement programs to ensure adherence to regulatory requirements.
- Conduct regular inspections and audits of manufacturers, sponsors, and distributors to maintain high standards.
- Engage and Inform Stakeholders
- Foster transparent and effective communication with healthcare professionals, consumers, and industry stakeholders.
- Provide timely and accurate information about therapeutic goods, regulatory decisions, and safety updates [4].
- Support Innovation and Access
Facilitate the introduction of innovative therapeutic goods to the market while maintaining rigorous safety and efficacy standards.
- Improve access to essential medicines and medical devices, particularly for underserved populations and rare diseases [5]
**Key Initiatives**
- Regulatory Science and Innovation
- Invest in regulatory science to develop new methodologies and tools for assessing therapeutic goods.
- Collaborate with research institutions and industry to support the development of innovative products.
- Digital Transformation
- Implement advanced IT systems and digital solutions to enhance regulatory processes and improve data management.
- Develop online platforms for stakeholders to access regulatory information and submit applications more efficiently.
- Global Collaboration
- Strengthen partnerships with international regulatory agencies to share information, harmonize standards, and address global health threats.
- Participate in international forums and initiatives to influence global regulatory practices [6].
- Stakeholder Engagement
- Conduct public consultations and stakeholder forums to gather input and feedback on regulatory policies and initiatives.
- Develop educational programs and resources to enhance stakeholder understanding of regulatory requirements.
- Regulatory Compliance and Integrity
- Enhance compliance monitoring and enforcement strategies to deter non-compliance and ensure high standards.
- Implement risk-based approaches to focus regulatory efforts on high-risk products and activities [7].
**Performance Measures**
- Timeliness: Measure the speed of regulatory processes, including application reviews and safety assessments.
- Quality: Assess the accuracy and thoroughness of evaluations, inspections, and enforcement actions.
- Stakeholder Satisfaction: Evaluate the satisfaction of stakeholders with TGA services and communication.
- Compliance Rates: Monitor compliance rates among manufacturers, sponsors, and distributors.
- Innovation Support: Track the number of innovative products approved and the efficiency of the approval process [8].
Regulation of Therapeutic Goods by TGA
The TGA regulates therapeutic goods throughout their lifecycle in number of ways like (Figure 1):
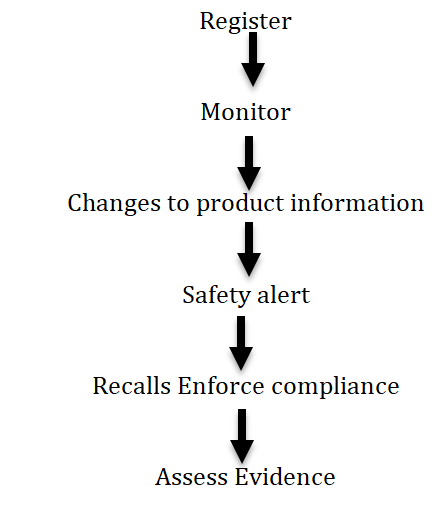
Pharmacovigilance and Special access branch
It is responsible for pre and post market monitoring compliances of medicines on the Australian register of therapeutic goods (ARTG). It includes monitoring of more than 27,354 medicines and it undertakes 18,000 ADR reports relating to medicines and vaccines 130 risk management plan evaluations and 60,0000notifications about clinical trials, authorised prescribers managed by experimental product section [9].
Risk management plan in Pharma covigilance
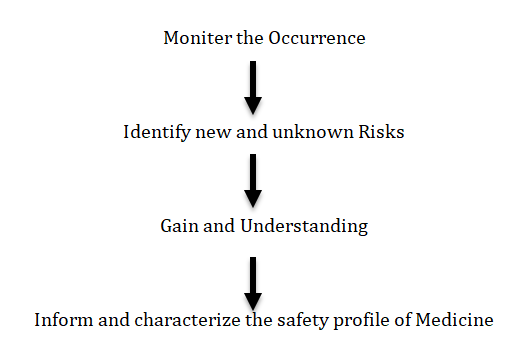
An RMP is a detail description of a risk management system which it contains description and analysis of safety profile about medication and includes a set of pharmacovigilance and risk minimisation activities. It covers entire life cycle of a medicine (Figure 2).
In pharmacovigilance (PV), a Risk Management Plan (RMP) is a critical tool used to ensure the safe use of medicinal products by identifying, assessing, and minimizing risks throughout the product’s lifecycle. The RMP in pharmacovigilance is designed to proactively manage known and potential risks associated with a drug or biological product, ensuring that the benefits of the product outweigh its risks [10].
Components of RMP
- Safety specification
- Summary of safety concerns
- Pharmacovigilance plan
- Risk minimisation plan
- Australian specific annexure.
Requirement of RMP
RMP is required for all the below applications
• New chemical entities Pre-Marketing Phase: During the development of an NCE, potential risks are identified through non-clinical studies and clinical trials. The RMP begins with a thorough risk analysis based on these studies. Post-Marketing Surveillance: Once an NCE is approved, RMPs guide post-marketing studies and Pharmacovigilance activities to monitor real-world safety. This is particularly important for rare side effects that might not be evident in pre-market trials. Risk Minimization Activities: These might include restricted distribution programs, patient education, and healthcare professional training to ensure the NCE is used safely and effectively. Regular Updates: The RMP is a dynamic document that evolves with new safety information. Regulatory authorities require regular updates to the RMP based on post-marketing data. Biosimilar medicines: An RMP may be required if the biosimilar does not have all of the same indications and presentations as the originator product, so the need for risk minimisation for safety concerns resulting from medication error can be considered [11].
• Vaccines Safety Monitoring: Vaccines are administered to large populations, including healthy individuals, so their safety profile is critical. RMPs are developed to monitor adverse events post-licensure, especially for rare or long-term effects that might not be detected in clinical trials. Risk Identification: Specific risks are identified during the pre-clinical and clinical development phases, such as potential allergic reactions, autoimmunity issues, or specific adverse events associated with vaccine adjuvants. Mitigation Strategies: RMPs for vaccines include strategies like additional studies, enhanced pharmacovigilance (e.g., active surveillance systems), and communication plans to inform healthcare professionals and the public. Benefit-Risk Assessment: The RMP ensures that the benefit-risk balance remains positive throughout the vaccine’s lifecycle by continuously updating the plan as new data emerge [12].
Class 3 and 4 biological products: Class 3 biologicals are typically moderately complex biologics with potential risks that require careful monitoring but are generally considered to have a favourable benefit-risk profile.
**RMP Application in Class 3 Biologicals**
Risk Identification and Characterization
Clinical and Preclinical Data: RMPs for Class 3 biologicals are developed based on data gathered from preclinical studies and clinical trials. These studies help identify potential immunogenicity, adverse reactions, or long-term safety concerns specific to biologics.
Targeted Risk Analysis: The RMP focuses on risks that are more likely to occur in specific patient populations, such as those with compromised immune systems or patients with comorbid conditions [12].
Pharmacovigilance Activities
Standard Post-Marketing Surveillance: RMPs for Class 3 biologics include routine post-marketing surveillance to monitor for adverse events. This is particularly important for detecting rare side effects that may not have been apparent in clinical trials.
Registry Studies: In some cases, patient registries are established to collect real-world data on the safety and effectiveness of the biologic in diverse populations [13].
Risk Minimization Measures
Labelling and Educational Materials: Clear labelling and educational materials are crucial to inform healthcare providers and patients about proper use, potential risks, and monitoring requirements.
Patient Monitoring: Depending on the biologic, the RMP may include guidelines for regular monitoring of patients, such as blood tests or other diagnostic measures to detect potential adverse effects early [14].
Benefit-Risk Evaluation
Ongoing Assessment: The benefit-risk profile of Class 3 biologics is continuously monitored and updated as new data emerge, ensuring that the biologic’s benefits continue to outweigh the risks in its intended use.
**Class 4 Biologicals**
Class 4 biologicals are typically the most complex and high-risk biologics, often including advanced therapies like gene therapies, cell therapies, or biologics with novel mechanisms of action. These products require stringent risk management strategies [13].
RMP Application in Class 4 Biologicals
Extensive Risk Identification and Characterization
Comprehensive Risk Assessment: The RMP for Class 4 biologics includes a thorough analysis of potential risks, including long-term safety concerns, immunogenicity, off-target effects, and other issues unique to advanced biologics.
Clinical Trial Data: RMPs are informed by detailed clinical trial data, with particular attention to any serious adverse events, delayed effects, or safety signals that may emerge.
Enhanced Pharmacovigilance
Intensive Post-Marketing Surveillance: Given the high-risk nature of Class 4 biologics, RMPs include enhanced pharmacovigilance measures such as active surveillance, risk registries, and long-term follow-up studies.
Risk-Benefit Monitoring: Continuous monitoring of the risk-benefit ratio is essential, especially as these products are used in broader populations or for longer periods.
Risk Minimization Strategies
Restricted Access Programs: For some high-risk biologics, access may be restricted to certain healthcare facilities or providers with specialized training to ensure the safe administration of the product.
Extensive Education and Training: Healthcare providers involved in the administration of Class 4 biologics receive extensive training, and patients may be provided with detailed information on potential risks and the importance of follow-up care.
Patient and Provider Agreements: In some cases, risk management may include agreements between patients and providers to ensure adherence to monitoring protocols and safe use practices [14].
Regulatory Requirements and Post-Approval Studies
Mandatory Post-Approval Studies: Regulatory authorities may require post-approval studies as part of the RMP to gather additional safety and efficacy data. These studies can help identify long-term effects or rare adverse events.
Regular Updates to the RMP: Given the complexity of Class 4 biologics, the RMP is regularly updated based on new data, including findings from ongoing studies, real-world evidence, and adverse event reports [15].
Australian Context
Registering a medicine in the European Union also requires an RMP.
Following things should be considered about risk management of medicines in Australian include:
- Indigenous population
- Large Asian population
- Rurality
- lack of specialist services
- Differences between state and federal control over some aspects of how medicines are used
- Risk management activities [16].
Pharmacovigilance plan
Risk minimization activities
- Risks are minimized by–including:
- The warnings/precautions/contraindications on the product information on packaging
- Educating parents and health professionals of specific risks
- Restricting access to particular prescriber or patient group
- Encouraging the reporting of ADR.
- Additional information to minimise the risk factors include:
- Product information
- Consumer medicine information
- Directions for use document
- Labelling, pack size and design
- Legal status
- Educational programmes
- Prescriber checklists
- DHCP
- Letters
- Controlled access programmes
- Medicinal software alerts [17].
RMP evaluation
• Evaluation can be done as a part of registration application.
- Recommendations can be suggested by evaluator to delegate who consider these considerations.
- The sponsor can take decision to respond to issues raised during the TGA.
- Evaluation team consists team compromises doctors, pharmacists and a toxicologist.
TGA Post market pharmacovigilance
Adverse Event Reporting and Monitoring Adverse Event Reporting System: The TGA operates the Database of Adverse Event Notifications (DAEN), where healthcare professionals, patients, and pharmaceutical companies can report adverse events associated with therapeutic goods. This database is essential for identifying new safety concerns and assessing the ongoing safety of products. Signal Detection and Assessment: The TGA uses the data from DAEN to perform signal detection, where patterns or trends in adverse events are analyzed to identify potential safety signals. These signals may indicate new or previously unrecognized risks associated with a product [18].
Risk Management Plans (RMPs) RMP Requirements: The TGA requires the submission of RMPs for new medicines and certain high-risk medicines. These plans outline how the risks associated with a product will be managed throughout its lifecycle. The TGA reviews these plans as part of the product’s approval process and continues to monitor the implementation of RMPs post- marketing. RMP Updates: As new safety data emerge, the TGA may require updates to the RMP to address any new risks or to enhance existing risk management strategies. The TGA monitors the effectiveness of RMPs through ongoing Pharmacovigilance activities.
Post-Market Reviews and Studies Post-Authorization Safety Studies (PASS): The TGA may request or mandate post-marketing studies to gather additional safety data on a product. These studies are often part of the RMP and are used to better understand the safety profile of a medicine in the real-world setting. Periodic Safety Update Reports (PSURs): Pharmaceutical companies are required to submit PSURs to the TGA, which provide a comprehensive update on the safety of a product, including new data and an analysis of the benefit-risk balance. The TGA reviews these reports to ensure ongoing safety [19].
Risk Minimization Activities Communication and Education: The TGA may implement or require pharmaceutical companies to undertake risk minimization activities such as issuing safety alerts, updating product labeling, and developing educational materials for healthcare providers and patients. Regulatory Actions: If a significant risk is identified, the TGA can take regulatory actions such as restricting the use of a product, requiring additional warnings on labels, or, in extreme cases, withdrawing the product from the market.
Collaboration and International Cooperation Global Pharmacovigilance Networks: The TGA collaborates with other regulatory agencies globally, such as the European Medicines Agency (EMA), the U.S. Food and Drug Administration (FDA), and the World Health Organization (WHO). This collaboration helps in sharing safety data, harmonizing pharmacovigilance practices, and responding to global safety issues. International Databases: The TGA contributes to and accesses international adverse event databases, such as Vigibase, maintained by the WHO, to monitor global safety trends and signals.
Transparency and Public Access to Information Public Database Access: The TGA provides public access to the DAEN, where information on adverse events reported in Australia is available. This transparency helps inform healthcare professionals and the public about potential risks associated with therapeutic goods. Safety Alerts and Updates: The TGA regularly publishes safety alerts, product recalls, and updates on its website, providing timely information about emerging safety concerns and regulatory actions taken to mitigate risks.
Regulatory Guidance and Industry Compliance Guidelines for Industry: The TGA provides detailed guidelines for pharmaceutical companies on pharmacovigilance requirements, including how to report adverse events, the preparation and submission of RMPs, and the conduct of post-marketing studies. Compliance Monitoring: The TGA monitors compliance with pharmacovigilance obligations and can take enforcement actions against companies that fail to meet these requirements, including fines, product recalls, or suspension of marketing authorization.
- Maintaining the ADR system
- Analysis AE data regularly
- Evaluating information from sponsors, literature, other regulators and WHO.
- Undertaking safety filters, safety reviews band risk benefits reviews.
- Communicating information to health professions and consumers.
- Taking regulatory action as needed,
- Issues tracked through a work flow database.
Volume of reports during post marketing surveillance by TGA
- In 2014, the TGA received over 18,000 ADR.
- Around 1800(-10%) were assessed as being causality unclear
- Not an adverse event
- Insufficient information to assess
- Reaction was not associated or extremely unlikely to be associated with the medicine [20].
Conclusion
The regulators’ perspective on Pharmacovigilance is centered on safeguarding public health by ensuring the safety and efficacy of medicinal products. Through rigorous monitoring, data analysis, and stakeholder collaboration, regulators strive to mitigate risks and enhance the overall benefit-risk profile of drugs. Continued advancements in technology and international cooperation are essential for the future success of Pharmacovigilance efforts.
From a regulator’s perspective, the Therapeutic Goods Administration (TGA) plays a critical role in ensuring the safety, efficacy, and quality of medicinal products throughout their lifecycle, encompassing both premarketing and post- marketing phases. During the premarketing phase, the TGA rigorously evaluates clinical trial data, safety profiles, and the potential risks associated with medicinal products to determine their suitability for market authorization. This phase focuses on identifying and mitigating risks before the product reaches the market.
In the post-marketing phase, the TGA’s pharmacovigilance efforts become crucial in monitoring real-world safety data, detecting new or rare adverse events, and ensuring that the risk-benefit balance remains favourable. Through the implementation and continuous monitoring of Risk Management Plans (RMPs), as well as the enforcement of reporting obligations, the TGA ensures that any emerging safety concerns are promptly addressed.
The TGA’s dual focus on premarketing evaluation and post-marketing surveillance underscores its commitment to public health and safety. By maintaining rigorous standards and adaptive regulatory practices, the TGA ensures that medicinal products are both safe for use and effective in delivering therapeutic benefits, thereby fostering trust and confidence in the healthcare system.
References
-
Peyvandi F, Garagiola I, Mannucci P (2021) Post- authorization pharmacovigilance for haemophilia in Europe and the USA: Independence and transparency are keys. Blood reviews 49: 100828.
-
Aronson J (2017) Post-marketing drug withdrawals: Pharmacovigilance success, regulatory problems. Therapies 725(5): 555-561.
-
Hunsel F, Gardarsdottir H, Boer A, Kant A (2019) Measuring the impact of pharmacovigilance activities, challenging but important. British Journal of Clinical Pharmacology 85(10): 2235-2237.
-
Wiktorowicz M, Lexchin J, Moscou K (2012) Pharmacovigilance in Europe and North America: divergent approaches. Social science & medicine 75(1): 165-170.
-
Hartford CG, Petchel KS, Mickail H, Perez-Gutthann S, Mchale M, et al. (2006) Pharmacovigilance during the Pre-Approval Phases. Drug Safety 29: 657-673.
-
King CE, Pratt NL, Craig N, Thai L, Wilson M, et al. (2020) Detecting Medicine Safety Signals Using Prescription Sequence Symmetry Analysis of a National Prescribing Data Set. Drug Safety 43: 787-795.
-
Arlett P (2020) Measuring the impact of risk communications: Robust analytical approaches are key.. British journal of clinical pharmacology 86(4): 635-636.
-
Lucas S, Ailani J, Smith T, Abdrabboh A, Xue F, et al. (2022) Pharmacovigilance: reporting requirements throughout a product’s lifecycle. Therapeutic Advances in Drug Safety: 13.
-
Torka M, Mintzes B, Bhasale A, Fabbri A, Perry L, et al. (2019) Secret safety warnings on medicines: A case study of information access requests. Pharmacoepidemiology and Drug Safety 28: 551-555.
-
Yadav S (2008) Status of adverse drug reaction monitoring and pharmacovigilance in selected countries. Indian Journal of Pharmacology 40(Suppl 1): S4-S9.
-
Blake KV, deVries CS, Arlett P, Kurz X, Fitt H (2012) Increasing scientific standards, independence and transparency in post‐authorisation studies: the role of the European Network of Centres for Pharmacoepidemiology and Pharmacovigilance. Pharmacoepidemiology and Drug Safety: 21(7): 690-696.
-
Gini R, Fournie X, Dolk H, Kurz X, Verpillat P, et al. (2019) The ENCePP Code of Conduct: A best practise for scientific independence and transparency in noninterventional postauthorisation studies. Pharmacoepidemiology and Drug Safety 28(4): 422-433.
-
Engel P, Almas M, Bruin M, Starzyk K, Blackburn S, et al. (2017) Lessons learned on the design and the conduct of Post‐Authorization Safety Studies: review of 3 years of PRAC oversight. British Journal of Clinical Pharmacology 83: 884-893.
-
Patel N, Kesselheim A, Darrow J (2023) Trust and Regulation: Assuring Scientific Independence in the FDA’s Emergency Use Authorization Process. Journal of health politics, policy and law 48 (5): 799-820.
-
Tafuri G, Trotta F, Leufkens H, Pani L (2013) Disclosure of grounds of European withdrawn and refused applications: a step forward on regulatory transparency. British journal of clinical pharmacology 75(4): 1149- 1151.
-
Klika C, Kaeding M, Schmalter J (2017) The EU Pharmacovigilance System and Adverse Drug Reaction Reporting in Practice: A Critical Assessment. European Journal of Risk Regulation 8: 772-778.
-
Alvarez Y, Hidalgo A, Maignen F, Slattery J (2010) Validation of Statistical Signal Detection Procedures in EudraVigilance Post-Authorization Data. Drug Safety 33: 475-487.
-
Courtois E, Tubert-Bitter P, Ahmed I (2021) New adaptive lasso approaches for variable selection in automated pharmacovigilance signal detection. BMC Medical Research Methodology 21: 271.
-
Ding Y, Markatou M, Ball R (2020) An evaluation of statistical approaches to postmarketing surveillance. Statistics in Medicine 39(7): 845-874.
-
Ji X, Cui G, Xu C, Hou J, Zhang Y, et al. (2022) Combining a Pharmacological Network Model with a Bayesian Signal Detection Algorithm to Improve the Detection of Adverse Drug Events. Frontiers in Pharmacology 12: 773135.
- Gallic and Citric Acid Present in the Peels of Tropical Fruits as an Alternative in the Fight against Cancer
- Treating the Forehead Lines with Combination of Forehead and Glabellar Botulinum Toxin Among Japanese Patients
- Clinical Evaluation of Patients Suffering from Breast Cancer & Determination of Treatment Therapies and Better Strategies Related to Breast Cancer
- Medieval Recipes by Al-Zahrāwī for Heart Palpitations Treatment
- Etiology and Prescription Errors of Myocardial Infarction in Different Health Care Systems of Azad Kashmir
- Early Diagnosis and Multidisciplinary Management of Turner Syndrome: A Paediatric Case Study