Usher Syndrome: A Case Report of Two Brothers
Purpose: To report a case of Usher’s syndrome in two brothers. Case Report: Two brothers, 24 and 26 years of age, presented to Chittagong eye infirmary and training complex with the complaints of gradual diminution of vision in both eyes, especially at night. Their ocular examination revealed best corrected visual acuity of 6/36 in right eye and 6/24 in left eye. Dilated fundus evaluation showed pigmentary retinopathy. Perimetry revealed peripheral visual field constriction. Their systemic examination revealed sensorineural hearing loss. These clinical findings supported the diagnosis of Usher syndrome Type 1. The patients were advised for low vision aids to improve their quality of life and counseling regarding the prognosis of the disease was done with the patient’s family members. Conclusion: Early detection and appropriate intervention will help preserve the residual vision.
Umme Salma Akbar1*, Shams Mohammed Noman2 and Shally Biswas3
Purpose: To report a case of Usher’s syndrome in two brothers.
Conclusion: Early detection and appropriate intervention will help preserve the residual vision. Keywords: Usher Syndrome; Pigmentary Retinopathy; Sensorineural Hearing Loss
Introduction
Usher syndrome is a rare genetic disorder characterized by retinitis pigmentosa and congenital sensorineural hearing loss. It was first described by Von Graefe A, et al. [1] in 1858 but was named for Charles Usher, a Scottish ophthalmologist who identified the disorder’s hereditary nature and recessive inheritance pattern [1]. Here we report the clinical ophthalmic presentation of two brothers with Usher syndrome Type I.
Case Report
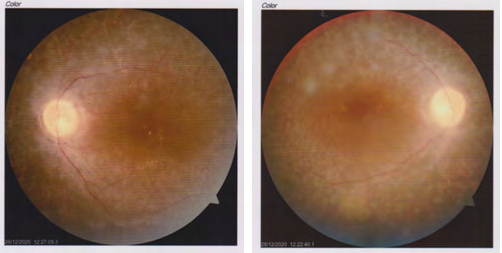
Two brothers, 24 and 26 years of age, presented to Chittagong eye infirmary and training complex, with the complaints of gradual diminution of vision in both eyes, especially at night. Their birth history revealed normal delivery at full term with hearing loss and inability to speak since childhood. A third sibling had no history of any visual or auditory problems. There was also no history of any similar condition in either of their parents or any other close relatives. On ophthalmological examination, they showed a best corrected visual acuity of 6/36 in the right eye and 6/24 in the left eye. The anterior segments were normal in both of them. Pupils were round, regular and reacting. Fundus examination revealed multiple bony spicules in the retinal periphery and marked arteriolar attenuation in both eyes of both brothers. The optic discs were both slightly pale (Figure 1). The intraocular pressures were 12mmHg in both eyes of both siblings. Perimetry revealed peripheral visual field constriction with relative defects in the paracentral region in both eyes. Upon consultation with an ENT specialist, audiometry revealed a profound hearing loss in both ears. The clinical findings supported the diagnosis of Usher syndrome Type I. Both the brothers were advised for low vision aids to improve their quality of life and counseling regarding the prognosis of the disease was done with the patient’s family members. The patients were advised regular follow up to the ophthalmologist as well as ENT specialist.
Discussion
Usher syndrome is a disorder characterized by retinitis pigmentosa and congenital sensorineural hearing loss. There are three different variants of Usher syndrome: Types I, II, III [2, 3]. Type I is the most severe form of the disorder with profound hearing loss and onset from birth accompanied by difficulty with night vision in the first decade of life. Type II is characterized by moderate hearing loss with later onset of night vision problems. Type III is accompanied by progressive hearing and night vision loss with onset in the late teenage years [2, 4, 5, 6]. Although differences in auditory and vestibular function are the distinguishing features, RP is the main ophthalmic manifestation shared by all three types [7]. In our case, the auditory loss was congenital and each child had difficulty with night vision beginning in early childhood. Therefore, these cases were categorized as Usher type I.
It affects both the auditory and visual systems and renders the patient functionally handicapped. It is important for children suffering from Usher syndrome to receive early diagnosis, which aids in re-adjustments on the part of the patients and parents for proper functioning in society [8]. Patients with Usher syndrome experience multiple visual disabilities, including reduced contrast sensitivity, poor dark adaptation, glare and night blindness. These disabilities may lead to difficulties with walking, recognizing objects and other activities of daily living. For night blindness and dark adaptation difficulties, a simple penlight is very useful. Near vision aids such as hand-held lighted magnifiers and closed- circuit televisions are helpful for reading and writing [7].
Appropriate genetic counseling to the parents can lead to awareness regarding the disorder and help to reduce its occurrence in future.
Conclusion
Early detection and appropriate intervention will help preserve the residual vision and strengthen auditory and speech potentials in these cases. The role of an Ophthalmologist in these cases includes diagnosis, treatment and interaction amongst other professionals on the treatment team.
Conflict of Interest
None
Acknowledgement
Chittagong Eye Infirmary and Training Complex
References
-
Von Graefe A (1858) Vereinzelte Beobachtungen and Bemerkungen: Exceptionelles Verhalten des Gesichtsfeldes bei Pigmententatung der Netzhaut. Von Graefes Arch Ophthalmol 4(2): 250-253.
-
Kremer H, Van Wijk E, Marker T, Wolfrum U, Ropeman R (2006) Usher syndrome: molecular links of pathogenesis, proteins and pathways. Hum Mol Genet 15: R262-R270.
-
Yan D, Liu XZ (2010) Genetics and pathological mechanisms of Usher syndrome. J Hum Genet 55(6): 327-335.
-
Rosenberg T, Haim M, Hauch AM, Parving A (1997) The prevalence of Usher syndrome and other retinal dyrtophy-hearing impairment associations. Clin Genet 51(5): 314-321.
-
Boughman JA, Vernon M, Shaver KA (1983) Usher syndrome: definition and estimate of prevalence from two high-risk populations. J Chronic Dis 36(8): 595-603.
-
Spandau UH, Rohrschneider K (2002) Prevalence and geographical distribution of Usher syndrome. Graefes Arch Clin Exp Ophthalmol 240(6): 495-498.
-
Sah RP, Gautam P, Shrestha JB, Joshi MR (2015) Usher syndrome: case reports of two siblings. Optom Vis perf 3(3): 174-177.
-
Young NM, Mets MB, Hain TC (1996) Early diagnosis of Usher syndrome in infants and children. Am J Otol 17(1): 30-34.
- Screening of Hospital Staff During World Glaucoma Week in a Tertiary Eye Care Centre
- Angioid Streaks with Macular Neovascularization: Clinical Insights from Two Cases
- Giant Kissing Naevus: An Oculoplastic Challenge
- Why Freedom of Vision Should Not Cost the Freedom of Feeling - LASIK in the Climate of Change
- Asymmetric Optic Nerve with Small Disc and Large Cup: A Rare and Challenging Case of Unilateral Optic Nerve Hypoplasia
- Large Angle Exotropia in a Child: A Case Report