A Rare Case of Evans Syndrome Associated With Optic Neuritis: A Case Report
Purpose: To report a case of Evans Syndrome associated with severe optic neuritis. Case Report: HAF, male, 26 years old, diagnosed with Evans Syndrome at the age of nine, with a history of headache and right eye pain with worsening movement. Fifteen days after the initial condition, visual clouding and decreased visual acuity began. Ophthalmological examination revealed visual acuity of 20/200 in the right eye with progressive worsening as the condition progressed; and 20/20 in the left eye. Visual field with absolute scotomata in the right eye and relative and absolute scotomatous points in all quadrants of the left eye. Gluorescein angiography findings included optic disc attenuation, without hyperfluorescent lesions. Conclusion: Evans Syndrome is a pathology that is gradually expanding in the field of clinical research, which has allowed new concepts related to pathogenesis and association with other comorbid conditions. In this case, we report the association with unilateral Optic Neuritis, responsible for progressive visual loss. Such knowledge aggregates the scientific community and translates into the need for an early multidisciplinary approach.
Introduction
Evans Syndrome (ES) was described for the first time by Robert Evans in 1951. It is a rare autoimmune hematological disease that occurs in a complex immunological context, characterized by the combination of two or more immune cytopenias, usually composed of autoimmune hemolytic anemia (AIHA) and immune thrombocytopenia [1, 2]. It is a disease with no predilection for gender, age or ethnicity. It has a chronic, relapsing and remitting course, is potentially fatal and requires immunosuppressive therapy with several different modalities [1, 3]. It can be divided into primary or idiopathic and secondary associated with connective tissue diseases (40%), hematological lymphoid diseases (30%) and primary immunodeficiency (18%). It is most often a diagnosis of exclusion [1, 3]. Comparative studies between therapeutic modalities are scarce, due to the rarity of the disease. Treatment can be performed with corticosteroids, intravenous immunoglobulin, immunobiologicals, immunosuppressant’s and even associated with splenectomy. Most recommendations are extrapolated and based on the treatment of immune thrombocytopenia and AIHA [3]. Ocular involvement is rare, but there are reports of sudden visual loss. In the literature, few articles were found that discussed the ocular involvement in this syndrome. A case of a child with ES with sterile inflammation of the Central Nervous System leading to diplopia along with neurological changes [4]. Another case in Japan of a patient with the syndrome who underwent vitrectomy for the treatment of vitreous hemorrhage and tractional retinal detachment [5]. Furthermore, a cause of visual loss in these patients, described in the literature, is vitreous hemorrhage due to severe thrombocytopenia. This work aims to report a case of Evans Syndrome associated with severe optic neuritis leading to a severe decrease in unilateral visual acuity and emphasize the importance of thinking about this diagnosis of exclusion in the face of autoimmune conditions that show signs of associated hematological disease. We emphasize the patient’s consent to voluntarily participate in the study and it only started after signing the free and informed consent form.
Case Presentation
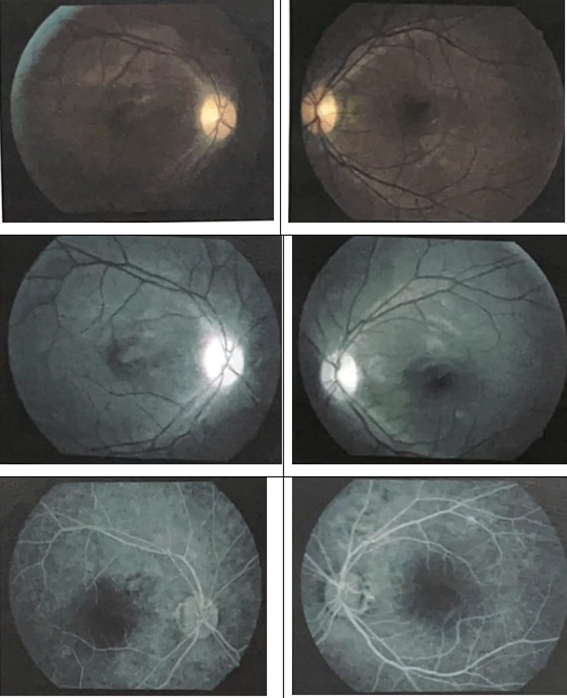
HAF, male, 26 years old, diagnosed with Evans Syndrome at the age of nine, with a history of headache onset and right eye pain that worsened on movement. Fifteen days after the initial condition, visual clouding and decreased visual acuity began. Ophthalmological examination revealed visual acuity of 20/200 in the right eye - with progressive worsening as the condition progressed; and 20/20 in the left eye. Afferent pupillary defect in the right eye, with Marcus Gunn’s pupils. He had preserved extraocular motility in both eyes. While the anterior segment and intraocular pressure were normal, the fundus examination of the right eye showed papilla pallor, optic nerve edges not visualized and diffuse reduction of the nerve fiber layer, magnetic resonance imaging of the orbits demonstrating sparse foci of T2 hypersignal/flair in the deep and subcortical white matter of the frontal and left anterior temporal lobes. An intravitreal injection of triamcinolone was attempted, followed by injectable hydrocortisone. No improvement of the condition with the mentioned treatment. Visual field with absolute scotomata in the right eye and relative and absolute scotomatous points in all quadrants of the left eye (Figure 1). Gluorescein angiography findings included optic disc attenuation, without hyper fluorescent lesions (Figure 2). Pulse therapy was also performed with methylprednisolone 1g/day for five days and plasmapheresis (five sessions) with little improvement in visual acuity and complete pain. Afterwards, the patient was indicated for monthly treatment with human immunoglobulin and semi-annual rituximab, even with the proposed treatment, he evolved with worsening visual acuity and only light perception vision.

Discussion
Evans Syndrome is an autoimmune hematological disorder characterized by the presence of two or more simultaneous or sequential cytopenias with positive Coombs. It is possible to frequently observe the association of AIHA and autoimmune thrombocytopenia, and neutropenia may also be present in a smaller number of cases (15-20%) [1, 3, 5, 6, 7, 8]. An annual incidence of 1-9/1,000,000 people is estimated. During adulthood, women are more affected, representing 60-70% of cases, while in childhood, it affects males in greater numbers, corroborating what was found in the case in question, since it is an individual from the male, diagnosed with the syndrome at the age of nine [1, 7]. Approximately 27 to 50% of ES cases are secondary, that is, they are triggered by infections, systemic autoimmune diseases, lymphoproliferative syndromes, hematopoietic stem cell transplantation and primary immunodeficiencies (in children they correspond to approximately 60% of ES cases). Some cases of SE have also been described after vaccines such as measles, mumps, rubella and influenza [1, 7, 8]. The pathophysiology of this syndrome is still unclear, but it is known that there is a defect in immune regulation that affects both humoral and cellular immunity. Among the changes observed are abnormalities of immunoglobulins, reduction of helper T cells and increase of suppressor T cells, ratio of abnormal Th1 and Th2 responses, production of autoantibodies by self-reactive plasmocytes, and several others [1, 3, 4, 8]. Patients with ES have periods of exacerbations interspersed with periods of remission. The
clinic is a consequence of thrombocytopenia and anemia, and the patient may evolve with bleeding, infections and thromboembolic events. Among the possible manifestations, petechiae, mucosal bleeding, fatigue, paleness, dyspnea, tachycardia, fever, jaundice, hepatosplenomegaly, hematuria, in addition to ophthalmological alterations in varying degrees of severity, such as ocular pain, visual clouding, reduction or loss of acuity can be observed. That was some of the symptoms presented by the evaluated individual [1].
The diagnosis of SE is one of exclusion described as AIHA, autoimmune thrombocytopenia and positive direct coombs that are not justified by another condition [1, 9]. AIHA is defined as a normocytic or macrocytic and normochromic hemolytic anemia associated with clinical jaundice and splenomegaly being justified due to the early destruction of red blood cells that occurs due to macrophage attack. This attack is suspected due to laboratory alterations that suggest it as reticulocytosis associated with elevation of indirect bilirubin, increased lactic dehydrogenase and reduction of haptoglobin. Direct coombs is positive in these patients because there is opsonisation of red blood cells, that is, an immune attack in which red blood cells are attached to antibodies. Autoimmune thrombocytopenia, on the other hand, usually has a rapid onset and is often associated with some other liver pathology, neoplasia, or bone marrow deficiency. The ophthalmological diagnosis of optic neuritis associated with this case initially consists of a condition of sudden visual loss in relatively young patients, more common between 18 and 45 years old, which occurs mostly unilaterally, being associated with ocular pain on extrinsic ocular movement in 90% of cases, in addition to visual loss for colors. It is possible to observe as an ophthalmological sign the presence of relative afferent pupillary defect in unilateral or asymmetrical cases, central, arched or altitudinal visual field defect. In 2/3 of the patients, there will be a normal optic disc with a retrobulbar neuritis but in 1/3 of these, there will be an edematous disc, the latter being more associated with children and adolescents [3].
For all patients, it is extremely important to request magnetic resonance imaging of the skull and orbits with gadolinium and fat suppression for follow-up and mainly therapeutic definition. In addition to performing a visual field test and excluding multiple sclerosis. Treatment, once multiple sclerosis and a previous history of optic neuritis have been excluded, depends directly on magnetic resonance imaging in which, once at least one typical demyelinating area is revealed, pulse therapy with steroids is started, ideally within the first 14 days of onset of ocular symptoms, methylprednisolone is recommended for 3 days, followed by oral prednisone 1mg/kg/day for 11 days, reducing to 20mg/day for 1 day, ending with 10mg/day for 3 days, being important to associate the entire period with antiulcer medication to gastric prophylaxis. If the MRI shows two or more typical demyelinating lesions, this patient needs, in addition to the aforementioned treatment, to be followed up by a neurologist and/or neuro-ophthalmologist to define the possibility of treatment with interferon-B-alpha, glatiramer acetate or fingolimod within 28 days [3].
With a negative MRI, the indication of treatment with steroids is only prescribed if the patient has previous visual impairment of the contralateral eye for some reason or when he requests treatment even though he has been advised of the risks. In patients with a diagnosis of multiple sclerosis or previous optic neuritis, pulse therapy should not be performed, just observe the evolution [3]. For the patient in the case, diagnosed with ES, in addition to the ophthalmological manifestations, the clinical manifestations of the disease such as hemolytic anemia, thrombocytopenia, splenomegaly should be treated with the treatments already established in the literature for these conditions [3, 9]. Reports of ophthalmological manifestations involving patients with ES are very scarce, which makes the therapeutic approach a challenge. Few involvements are described, such as vitreous hemorrhage and retinal detachment associated with the syndrome. Among the neurological lesions associated with ES are demyelinating polyneuropathy and lymphoplasmocellular encephalitis, with rare manifestations and descriptions in the literature. No cases of optic neuropathy were found in patients with ES in the literature review of this work.
Conclusion
Evans Syndrome is a rare pathology with gradual expansion in the field of clinical research, which has allowed new concepts, related to pathogenesis and association with other comorbid conditions. However, the scarcity of studies associating ophthalmological alterations associated with the syndrome makes diagnosis and therapeutic study difficult. In this case, we report the association with unilateral Optic Neuritis, responsible for severe progressive visual loss. Such knowledge aggregates the scientific community and translates into the need for an early multidisciplinary approach.
Conflict of Interest
None. References
1. Vera MS, Gonzalez MDJJ, Moreno AS, Serrano OFP, Perez MJP (2022)Síndrome de Evans. Rev Med Clin 22(4): 207- 212.
2. Evans RS, Takahashi K, Duane RT, Payne R, Liu C (1951)
Primary thrombocytopenic purpura and acquired hemolytic anemia. AMA Arch Intern Med 87(1): 48-65.
3. Mantadakis E, Farmaki E (2017) Natural History, Pathogenesis, and Treatment of Evans Syndrome in Children. J Pediatr Hematol Oncol 39(6): 413-419.
4. Simon OJ, Kuhlmann T, Bittner S, Tidow CM, Weigt J, et al. (2013) Evans syndrome associated with sterile inflammation of the central nervous system: A case report. J Med Case Rep 7(1): 262.
5. Ohnishi A, Kaneko M, Hori S, Teramura M, Kato S (2000) Vitrectomy for Vitreous Bleeding and Tractional Retinal Detachment in a Case of Evans Syndrome. Jpn J Ophthalmol 44(2): 177-179.
6. Larquin I, Gladys D, Risco M, Ii A, Yanet A, et al. (2008) Sindrome de Evans: Reporte de un caso. Rev Arch Médico Camagüey 12(1).
7. Fattizzo B, Michel M, Giannotta JA, Hansen DL, Arguello M, et al. (2021) Evans syndrome in adults: an observational multicenter study. Blood Adv 5(24): 5468-5478.
8. Hazmi AA, Winters M (2019) Evans Syndrome. Clin Pract Cases Emerg Med 3(2): 128-131.
9. Mathew P, Chen G, Wang W (1997) Evans Syndrome. J Pediatr Hematol Oncol 19(5): 433-437.
- Screening of Hospital Staff During World Glaucoma Week in a Tertiary Eye Care Centre
- Angioid Streaks with Macular Neovascularization: Clinical Insights from Two Cases
- Giant Kissing Naevus: An Oculoplastic Challenge
- Why Freedom of Vision Should Not Cost the Freedom of Feeling - LASIK in the Climate of Change
- Asymmetric Optic Nerve with Small Disc and Large Cup: A Rare and Challenging Case of Unilateral Optic Nerve Hypoplasia
- Large Angle Exotropia in a Child: A Case Report