Protein Kinase C-Dependent Phosphorylation of Human Pleckstrin Reduces its Capacity for Self-Association and Impairs its Ability to Bind Phosphoinositide
Pleckstrin is a major substrate of protein kinase C in lymphocytes, macrophages, monocytes, granulocytes and platelets. In these cells, pleckstrin is expressed at high levels and represents approximately 1 % of total cellular protein. Pleckstrin plays an important role in protein kinase C-dependent secretion, and abberant pleckstrin phosphorylation has been associated with disease. The mechanism, however, by which phosphorylation regulates pleckstrin function is unknown. Here, we show that native pleckstrin self-associates to form dimers that reduce its binding capacity to phosphoinositides. Phosphomimetic amino acid substitutions at residues normally phosphorylated by protein kinase C result in significantly reduced pleckstrin dimerization. Although phosphomimetic forms of pleckstrin exhibit subtle conformational changes they retain most of their phosphoinositide binding properties. These findings suggest that phosphorylation regulates pleckstrin interaction with membranes via a dimerization-dependent mechanism.
Introduction
Pleckstrin is one of the most abundantly phosphorylated proteins following platelet activation [1, 2, 3]. Pleckstrin phosphorylation is mediated by protein kinase C (PKC) and pleckstrin is a major PKC substrate in platelets, granulocytes, monocytes, macrophages, and lymphocytes [4] where it represents about 1% of total protein. Pleckstrin is a modular protein composed of three domains of approximately equal size, each comprised of approximately 100 amino acids. The domains include N- and C-terminal pleckstrin homology (PH) domains, which flank a central disheveled eg10 and pleckstrin (DEP) domain. Like PH domains found in many other proteins, the PH domains of pleckstrin mediate membrane localization through phosphoinositide binding and regulate various protein-protein interactions [5, 6, 7, 8, 9, 10, 11].
The region between the N- terminal PH domain (NPH) and the DEP domain contains three residues (S113, T114 and S117) that are phosphorylated by PKC [12, 13]. The consequences of pleckstrin phosphorylation are poorly understood but are thought to influence pleckstrin structure and perhaps membrane localization, leading to cytoskeletal reorganization, cell-cell adhesion, and migration [14, 15, 16, 17, 18]. Aberrant pleckstrin expression and phosphorylation have been associated with chronic inflammation associated with periodontitis, rheumatoid arthritis, and cardiovascular diseases [19, 20], as well as bleeding disorders and poorly controlled diabetes [21, 22, 23, 24]. Prior studies found that pleckstrin is involved in exocytosis. In mononuclear phagocytes of poorly controlled diabetics pleckstrin was found to be hyperphosphorylated. Reduction of pleckstrin expression using siRNA caused a decrease in the secretion of proinflammatory cytokines [24]. Similarly, a study by Lian, et al. [17], using a pleckstrin-null mouse model, demonstrated that pleckstrin is an essential component of PKC-mediated granule exocytosis in platelets. Collectively, these studies provide strong evidence suggesting pleckstrin plays an important role in PKC- dependent exocytosis.
While the functional role of pleckstrin is becoming clearer, the mechanism underlying how phosphorylation regulates pleckstrin remains unanswered. Numerous phosphorylation-dependent mechanisms for regulation have been reported for a wide range of proteins. With respect to pleckstrin, phosphorylation has been proposed to alter both its oligomeric state and its conformation, although evidence to support such mechanisms has not been provided [14, 25, 26, 27, 28, 29]. To better define the molecular mechanism through which phosphorylation regulates pleckstrin we have investigated the structural and functional consequences of pleckstrin phosphorylation.
Materials and Methods
Expression and Purification of Pleckstrin Proteins
All pleckstrin proteins were expressed and purified as hexahistidine fusion proteins in Escherichia coli BL21 (DE3) using the pDEST17 expression vector (Invitrogen). Bacteria were grown in standard LB medium supplemented with 10 mg ml-1 ampicillin at 37°C with shaking (225 rev min-1) until the absorbance at 600 nm reached 0.5. Protein expression was induced using 1.0 mM IPTG and the incubation temperature was lowered to 20°C. After a 5-hour induction period, bacteria were harvested by centrifugation at 3 315 x G for 10 minutes at 4°C. Each 1L pellet was then resuspended in 8 ml of 1x phosphate buffered saline (PBS) and centrifuged at 3 315 x G for 10 minutes at 4°C. The resulting cell pellets were then flash frozen in liquid nitrogen and stored at -80°C. Prior to lysis using a French press, pellets (2L) were resuspended to a final volume of 35 ml using nickel A buffer (20 mM Tris-HCl, pH 7.5, 1.5 M KCl, 0.06% LDAO and 5 mM imidazole). After lysis, samples were subjected to centrifugation at 48 384 x G for 40 minutes at 4°C and the supernatants were applied to a HiTrap nickel affinity column (GE Healthcare). Bound proteins were eluted using 500 mM imidazole following sequential washes with 5 and 15 mM imidazole, which was accomplished by mixing appropriate volumes of nickel A buffer and nickel B buffer (20 mM Tris-HCl pH 7.5, 1.5 M KCl, 0.06% LDAO and 500 mM imidazole). After incubation with 50 mM EDTA for 60 minutes at 4°C to remove trace amounts of Ni2+, protein was buffer exchanged into 20 mM Tris-HCl pH 7.5 and 300 mM KCl using a HiPrep 26/10 desalting column (GE Healthcare). The hexahistidine tag was removed by cleavage with TEV protease. This sample was then diluted to 200 mM KCl using S-A buffer (20 mM Tris-HCl 7.5) and applied to a HiTrap SP Sepharose HP ion exchange column (GE Healthcare) to further purify the protein from any remaining contaminants. The protein of interest was subsequently eluted using a salt gradient generated by the application of S-B buffer (20 mM Tris-HCl 7.5 and 1 M KCl). Final protein samples were buffer exchanged into experiment specific buffers (see below) and concentrated using ultrafiltration (GE Healthcare). Protein purity was consistently greater than 95% as determined by SDS-PAGE analysis.
PIP Strip and PIP Array Binding Assays
PIP StripTM and PIP ArrayTM membranes were obtained from Echelon Biosciences Inc. Assays were performed using the following protocol. Membranes were blocked with PBS + 1% nonfat-dry milk for 1 hour at 21.0°C prior to incubation with protein of interest at a concentration of 9.0x10-2µM in sample buffer (20 mM Tris-HCl pH 7.5 and 150 mM KCl) with gentle agitation at 21.0°C for 1 hour. Experiments involving dimeric pleckstrin were conducted at a concentration of 230µM. After discarding the protein solution membranes were washed with 15 ml of wash buffer (20 mM Tris-HCl pH 7.5, 150 mM KCl and 1% Tween-20) two times with agitation at 21°C for 15 minutes each. Membranes were then incubated for 1 hour at 21.0°C with anti-pleckstrin monoclonal antibody (Abnova) diluted 1:3500 in sample buffer. The primary antibody was discarded, and the membrane was washed as described above. Membranes were then incubated for 1 hour at 21.0°C with alkaline phosphatase coupled secondary antibody (Biorad) diluted 1:3300 in sample buffer. The membrane was then washed and incubated in 10 ml of developing solution (Biorad) for 15 mins. The membrane was then rinsed with water, air dried and scanned using a standard desktop computer scanner.
Gel Filtration Analysis
All gel filtration experiments were performed using a Superdex 200 10/300 GL gel filtration column (Amersham Biosciences) at 4.0°C. Protein samples were diluted in 20 mM Tris-HCl pH 7.5, 200 mM KCl and 2 mM TCEP. The same buffer was used to equilibrate the column with at least 10 column volumes prior to sample injection. All samples were injected at a flow rate of 0.1 ml/min. Following sample injection, the flow rate was adjusted to 0.5 ml/min for the duration of the experiment. Molecular weights of the samples were calculated from a standard curve generated using known protein standards (Amersham Biosciences) that were used to calibrate the column (shown in Figure S1 of the supplemental material).
Analytical Ultracentrifugation Sedimentation Equilibrium
Sedimentation equilibrium experiments were conducted at 4.0°C on a Beckman Optima XL-A analytical ultracentrifuge. Pleckstrin and pseudophosphorylated pleckstrin were studied at loading concentrations ranging from 86 to 7.6µM and 150 to 13.5µM, respectively. 2-channel, 3 mm path length cells were used for concentrations above 40µM (35µL), otherwise samples were loaded into 6-channel, 12 mm path length cells (135—µL). Data were acquired at 12, 16, 20 and 24 krpm, as an average of 4 absorbance measurements at 250 and 280 nm using a radial spacing of 0.001 cm. Sedimentation equilibrium at each rotor speed was achieved within 48 hours. Data for each sample were analyzed globally in terms of a monomer-dimer self-association in SEDPHAT 7.03 (Schuck, 2003) with the implementation of mass conservation. The solution density ρ and viscosity ᵑ protein partial specific volumes v and extinction coefficients at 280 nm were calculated in SEDNTERP 1.09 [30]. Errors were determined using the method of F-statistics with a confidence level of 68.3%.
Analytical Ultracentrifugation Sedimentation Velocity
Sedimentation velocity experiments were conducted at 20.0°C on a Beckman Coulter Proteome XL-I analytical ultracentrifuge using both absorbance (280 nm) and Rayleigh interference optical detection systems. Pleckstrin and pseudophosphorylated pleckstrin were studied at loading concentrations ranging from 351 to 4.0µM and 185 to 10µM, respectively. 2-channel, 3 mm path length sector shaped cells were used for concentrations above 35µM (100µL), otherwise samples were loaded into 2-channel, 12 mm path length sector shaped cells (400µL). 100 scans were acquired at 7-minute intervals and rotor speeds of 50 krpm; absorbance data were collected as single absorbance measurements using a radial spacing of 0.003 cm. Data were analyzed in SEDFIT 11.9b [31] in terms of a continuous c(s) distribution using an s range of 0.5 to 10 with a linear resolution of 100 and confidence levels (F-ratio) of 0.68 and 0.85 for the absorbance and interference data, respectively. In all cases, excellent fits were observed with root mean square deviations ranging from 0.0040 – 0.0064 absorbance units and 0.0026 – 0.0103 fringes. Weighted average sedimentation coefficients, combining contributions from the pleckstrin monomer and dimer, were obtained from the integral of the c(s) distributions, and modeled in SEDPHAT 11.9b [31, 32] in terms of a monomer-dimer self-association. Errors in the dissociation constant were determined using the method of F- statistics with a confidence level of 68.3%. Partial proteolysis: Partial proteolysis experiments were conducted using trypsin (1 mg/ml in 1 mM HCl) diluted 1/10 into 20 mM Tris-HCl pH 7.5 and 1 mM EDTA. Pleckstrin proteins were in 20 mM Tris-HCl pH 7.5 and 100 mM KCl. Each 400µl reaction contained 340µl pleckstrin protein (final concentration 7.5µM), 20µl 50 mM MgCl2 (final concentration 2.5 mM) and 40µl trypsin (final concentration 0.4µM). The reactions were incubated at 21.0°C with 20µl aliquots being removed at 5-minute intervals and immediately mixed with 20µl of 2x SDS-PAGE loading dye. The samples were analyzed by SDS-PAGE on 15% gels.
Results
Native Pleckstrin Self-Associates to Form Dimers
Gel filtration and analytical ultracentrifugation experiments were performed to establish the oligomeric state of pleckstrin. To mimic phosphorylation, the three residues phosphorylated by protein kinase C (S113, T114 and S117) were mutated to glutamic acid residues. This mutant, referred to as pseudophosphorylated pleckstrin, has been used by other groups and is known to mimic phosphorylated pleckstrin [15, 33, 34]. Initially, both pleckstrin proteins were analyzed by gel filtration using a range of concentrations (25, 125 and 250µM). Pleckstrin has a theoretical molecular weight of 40.2 kDa. As shown in Figure 1, panel A, at a concentration of 25µM, native pleckstrin eluted in a volume corresponding to an apparent molecular weight of 44 kDa. When the concentration of native pleckstrin was increased to 125 or 250µM, the elution volumes decreased to corresponding molecular weights of 49 and 58 kDa respectively. Conversely, pseudophosphorylated pleckstrin eluted with a volume corresponding to 44 kDa at all concentrations tested. The observed concentration dependent shift in elution volumes suggested that native pleckstrin self-associates to form higher order oligomers. To further characterize and quantitate this apparent equilibrium the system was examined using analytical ultracentrifugation.
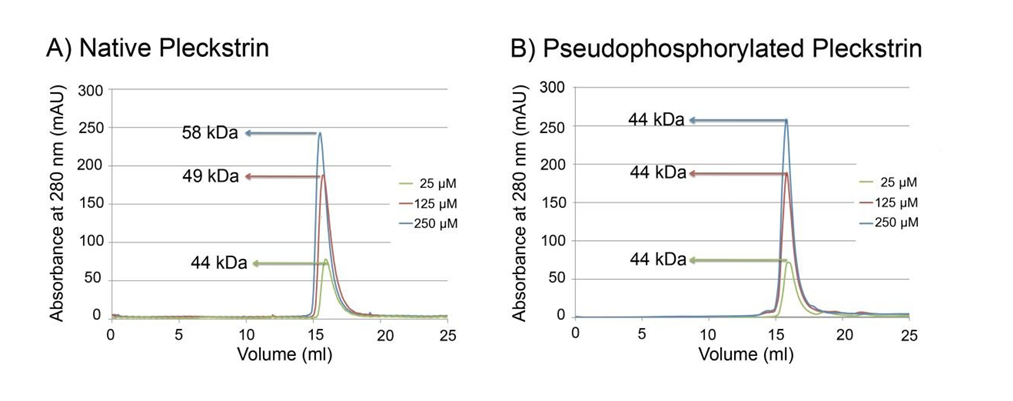
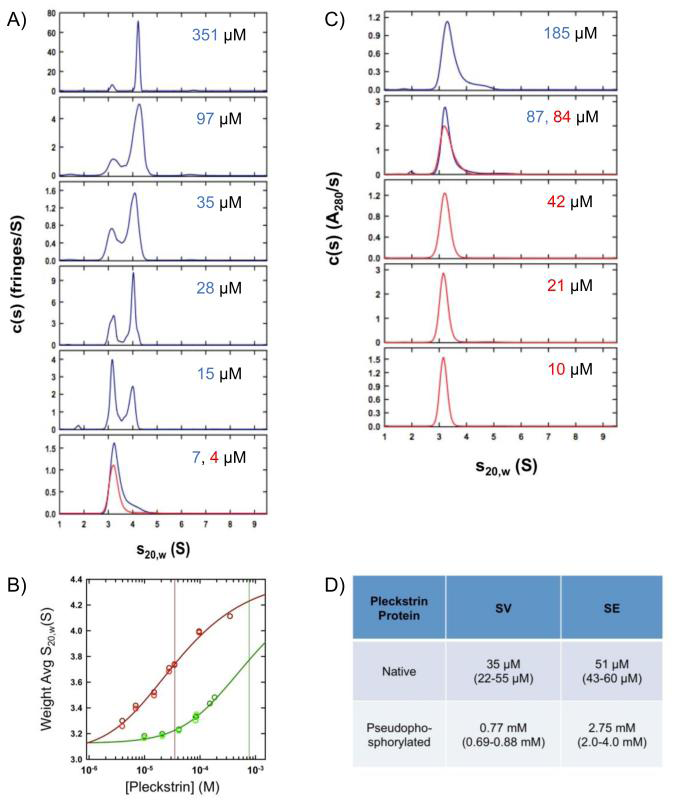
Figure 2: Native pleckstrin self-associates to form dimers with a higher affinity than pseudophosphorylated pleckstrin. Sedimentation velocity experiments on A) native and C) pseudophosphorylated pleckstrin. The c(s) distributions obtained using the program SEDFIT based on sedimentation velocity interference data collected at 50 krpm and 20.0°C for pleckstrin at denoted loading concentrations. 3 mm path-length cells were used for 351, 97, 35 µM native samples and 185, 87, 84, 42 µM pseudophosphorylated samples. Data sets in red and blue correspond to different protein preparations. B) Isotherms of weighted average sedimentation coefficients obtained from peak integration of the c(s) data. Data obtained for native pleckstrin are shown in red, whereas data obtained for the pseudophosphorylated mutant are shown in green. Lighter colored data points are obtained using absorbance optics whereas darker ones are based on interference optics. For each pleckstrin sample, data were fit globally to a monomer- dimer model (solid lines) with the best-fit dissociation constants shown as solid vertical lines. D) Table summarizing the sedimentation equilibrium data. Error ranges are given in brackets.
For native pleckstrin, weighted average sedimentation coefficients calculated by integration of the c(s) distributions obtained by sedimentation velocity (Figure 2A) were well modeled in terms of a monomer-dimer isotherm returning a Kd of 35µM (68% confidence interval 22 – 55µM), Figure 2B. At the lower loading concentrations, the slower sedimenting species (3.20 S) is estimated to have a molecular mass of 38 kDa, corresponding to a pleckstrin monomer. At the highest loading concentration (351µM), the major species (4.2 S) would appear to be a dimer with an estimated mass of 78 kDa. These observations were confirmed by sedimentation equilibrium, which returned a comparable monomer-dimer Kd of 51µM (68% confidence interval 43 – 60µM), (Figure 2D, S2). A similar set of experiments was carried out on pseudophosphorylated pleckstrin. No significant amount of dimer formation was observed at the highest concentration tested (185 µM), indicative of a weaker self-association (Figure 2B).
This was confirmed by the weighted average sedimentation coefficient isotherm, which returned a best- fit Kd of 0.77 mM (68% confidence interval 0.69 – 0.88 mM) (Figure 2B), along with a sedimentation coefficient of 3.19 S for the monomer. In this analysis, the sedimentation coefficient of the dimer was fixed to 4.50 S. Similar observations were made by sedimentation equilibrium in which a Kd of 2.75 mM (68% confidence interval 2.0 – 4.0 mM) was determined (Figure 2D, S3). Due to the low proportion of mutant dimer, it should be noted that values of the monomer-dimer Kd obtained by these methods represent estimates rather than precise values. Nonetheless, these data clearly demonstrate that the pseudophosphorylation mutation decreases dimerization affinity by at least 20- fold.
Pseudophosphorylation Alters the Conformation of Pleckstrin
Given the preliminary evidence suggesting that phosphorylation causes a conformational change in pleckstrin [25, 26, 27, 28, 29], this mode of regulation was also examined. Intrinsic fluorescence and CD spectroscopy experiments were unable to detect a conformational change between native and pseudophosphorylated pleckstrin (data not shown). Analysis by partial proteolysis yielded distinct digestion patterns suggestive of a phosphorylation dependent conformational change. As shown in Figure 3, the first difference observed between proteolysis of native and pseudophosphorylated pleckstrin is the rate at which the full-length proteins are degraded. Native pleckstrin is more resistant to trypsin digestion than pseudophosphorylated pleckstrin as evident by persistence of full-length native pleckstrin. The second difference is the presence of unique species in the digestion patterns of both native and pseudophosphorylated pleckstrin (highlighted by arrows in Figure 3). The observed differences in digestion patterns between native and pseudophosphorylated pleckstrin are consistent with a phosphorylation-dependent conformational change.
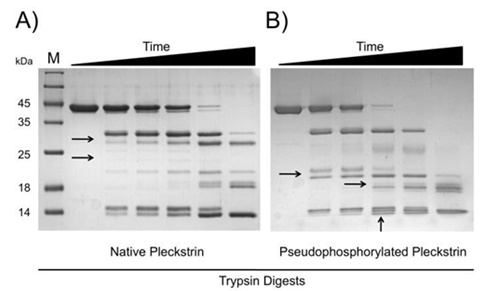
Figure 3: Pseudophosphorylation causes a conformational change in pleckstrin. Trypsin digests of native and pseudophosphorylated pleckstrin shown in panels A and B, respectively. Time points include 0, 5, 10, 15, 20 and 25 minutes. Black arrows highlight unique species. Samples were analyzed by 15% SDS-PAGE and stained with Coomassie brilliant blue.
Effect of Pseudophosphorylation on Pleckstrin Phosphoinositide Binding Properties
Having investigated the structural consequences of pleckstrin phosphorylation, we next explored possible functional consequences by analyzing phosphoinositide binding. While the phosphoinositide binding properties of individual PH domains have been examined [9, 10] the binding properties of full-length pleckstrin have yet to be established. Phosphoinositide binding specificities of both native and pseudophosphorylated pleckstrin proteins were determined at concentrations where the proteins are predominately in a monomeric state. Using qualitative lipid overlay assays, both forms of pleckstrin were found to bind specifically to phosphorylated phosphoinositides with only very limited affinity for other lipids (Figure 4A and B). The
one exception to this trend was that both proteins bound to phosphatidic acid, albeit with weaker affinity compared to phosphoinositides. To gain a better understanding of the relative binding affinities for individual phosphoinositides, both pleckstrin proteins were incubated with membranes containing varying amounts of each phosphoinositide (Figure 4, panels C and D). Based on this analysis it appeared that both native and pseudophosphorylated pleckstrin bind PtdIns (3,4,5) P3 with highest affinity followed by PtdIns (3,4) P2 and PtdIns (3) P.
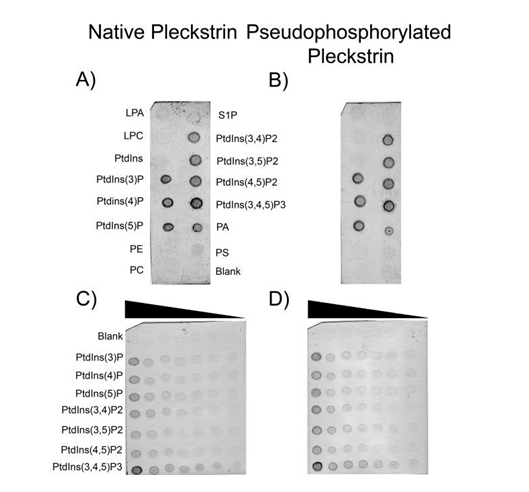
Figure 4: Protein-lipid overlay assays of native and pseudophosphorylated pleckstrin. Panels A and B show PIP Strip results for native and pseudophosphorylated pleckstrin (9.0x10-2µM final concentration) respectively. Each spot contains 100 pmol of lipid. Panels C and D are PIP Arrays where varying concentrations of lipid are blotted (100, 50, 25, 12.5, 6.25, 3.13, and 1.58 pmol). LPA, lysophosphatidic acid; S1P, sphingosine-1-phosphate; LPC, lysophosphocholine; PE, phosphatidylethanolamine; PC phosphatidylcholine; PA, phosphatidic acid; PS, phosphatidylserine; PtdIns, phophatidylinositol (with numbers in brackets indicating position of phosphate groups).
Pleckstrin Dimerization Blocks Phosphoinositide Binding
Having shown that phosphorylation alters pleckstrin’s oligomeric state, we sought to determine the effect of dimerization on pleckstrin’s phosphoinositide binding properties. To accomplish this native pleckstrin was analyzed in lipid overlay assays at a concentration favoring dimerization (230µM). As shown in Figure 5, dimerization significantly
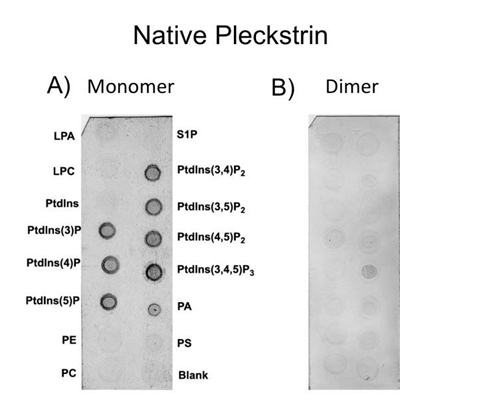
Figure 5: Protein-lipid overlay assay comparing native pleckstrin as a monomer and dimer. Panel A shows binding of native pleckstrin at a concentration favoring the monomeric form (9.0x10-2µM). Panel B is native pleckstrin at a concentration favoring the dimeric form (230µM). Each spot contains 100 pmol of lipid. LPA, lysophosphatidic acid; S1P, sphingosine-1- phosphate; LPC, lysophosphocholine; PE, phosphatidylethanolamine; PC phosphatidylcholine; PA, phosphatidic acid; PS, phosphatidylserine; PtdIns, phophatidylinositol (with numbers in brackets indicating position of phosphate groups).
decreased the binding of pleckstrin to phosphoinositides (compare panels A and B). Unlike monomeric pleckstrin, which bound promiscuously to all seven phosphoinositides, dimeric pleckstrin bound only weakly to PtdIns (3,4,5) P3. To rule out the possibility that the antibody-binding site is occluded by the dimerization interface we performed an identical experiment using a different, polyclonal pleckstrin antibody and observed the same result.
Discussion
Phosphorylation of Pleckstrin Governs Self- Association
Native pleckstrin dimerizes with a Kd of 51 µM and pseudophosphorylation, which is known to mimic phosphorylation of pleckstrin, decreases dimerization affinity by approximately 20-fold. Given that the intracellular concentration of pleckstrin is approximately 15µM (24) it is reasonable to expect that a proportion of native pleckstrin will exist as a dimer in vivo. This result is corroborated by previous, in vivo chemical cross-linking studies that were able to detect pleckstrin dimers in resting platelets (McDermott and Haslam, 1996). When platelets were stimulated prior to cross-linking, pleckstrin dimers were not detected. This observation is consistent with the evidence presented here suggesting that phosphorylation decreases the dimerization affinity by at least 20-fold. The biochemical analysis reported here combined with in vivo results from previous chemical cross-linking studies provides compelling evidence for a model in which phosphorylation regulates pleckstrin by altering its oligomeric state.
Phosphomemetic Substitutions Induce a Conformational Change in Pleckstrin
In addition to showing that pleckstrin pseudophosphorylation alters its oligomeric state we found evidence for an associated conformational change based on observed differences in proteolytic digestion patterns. These differences include the presence of unique species and an altered rate of proteolysis. Whereas full- length, pseudophosphorylated pleckstrin is degraded more rapidly with no full- length protein remaining after 15 minutes, native pleckstrin is more resistant to digestion persisting for over 20 minutes. The digestion patterns of both native and pseudophosphorylated pleckstrin also contain unique species. This observation is important since partial proteolysis was conducted under conditions where both forms of pleckstrin are predominantly monomeric. Although the presence of residual dimers may affect the rate of proteolysis, dimers are unlikely to result in the generation of unique species. The observed differences are consistent with pseudophosphorylation exposing one or more cleavage sites that were only partially exposed in native pleckstrin. We suggest that this conformational change is rather subtle based on the small differences in sedimentation coefficients for monomeric native and pseudophosphorylated pleckstrin, as well as the inability to detect changes in the intrinsic fluorescence and CD spectra. Data supporting a phosphorylation-dependent conformational change in pleckstrin is further strengthened when combined with findings from a prior study where pleckstrin was used for the creation of a probe to detect PKC phosphorylation. To create the probe, the C-terminal PH domain of pleckstrin was removed and two additional fluorescent domains were attached to the N- and C-termini of truncated pleckstrin [25, 26, 29]. The three residues phosphorylated by PKC (S113, T114 and S117) were retained in a loop region between the N-terminal PH and DEP domains [12, 13]. The probe’s mechanism of action is based on a change in the relative position of the two fluorescent domains with respect to one another following phosphorylation. A change in fluorescence was attributed to a change in the relative positions of the NPH and DEP domains; a concept that was further supported by preliminary NMR data. In the context of full- length pleckstrin it is likely that the conformational change induced by phosphorylation alters the position of NPH with respect to the rest of pleckstrin. Crystal structures of native and pseudophosphorylated pleckstrin may provide valuable information regarding the precise nature of the resulting conformational change.
Self-Association of Pleckstrin Prevents Phosphoinositide Binding
In order to understand how phosphorylation- dependent structural changes might affect function, the phosphoinositide binding properties of pleckstrin were examined. Binding properties of both pleckstrin proteins were examined using concentrations favoring their monomeric forms. Protein- lipid overlay assays revealed that native and pseudophosphorylated pleckstrin bind specifically to phosphoinositides. Among the different phosphoinositides however, both proteins appeared to be promiscuous, binding to all seven phosphoinositides. Furthermore, pseudophosphorylated pleckstrin did not demonstrate a significantly enhanced specificity towards a specific phosphoinositide (PtdIns (3) P being a lone exception) that convincingly identifies a single phosphoinositide ligand compared to native pleckstrin. These findings suggest that phosphorylation does not alter the binding properties of pleckstrin in such a way that enhances its specificity so that a single phosphoinositide is clearly preferred.
While both native and pseudophosphorylated pleckstrin bound readily to all phosphoinositides under concentrations favoring monomer, binding of native pleckstrin was almost completely abolished at higher concentrations favoring a dimeric form. This finding is both consistent with and helps explain previous observations in studies of pleckstrin in platelets and neutrophils. In resting cells pleckstrin is not phosphorylated and was found to be predominately cytosolic. Following cellular stimulation, pleckstrin became phosphorylated and translocated to the membrane. Given the available evidence it appears that non-phosphorylated pleckstrin exists in a monomer-dimer equilibrium, which serves to prevent membrane localization. Phosphorylation causes a shift to the monomeric form thereby exposing phosphoinositide binding sites and allowing pleckstrin to interact with phosphoinositides in the membrane. As discussed previously, phosphorylation has been proposed to cause changes in both the oligomeric state and the conformation of pleckstrin. Based on the results presented here we propose a model describing how phosphorylation regulates pleckstrin. In this model, native pleckstrin exists as a cytosolic protein in amonomer-dimer equilibrium (Kd 51± 9±µM). Dimerization impairs phosphoinositide binding thereby preventing membrane localization. Phosphorylation by PKC results in a subtle conformational change and equilibrium shift from dimer to monomer. The shift in equilibrium relieves the impairment of phosphoinositide binding allowing pleckstrin to localize to the membrane. Once at the membrane, phosphorylated pleckstrin appears to play a role in secretion through a currently unknown mechanism [35].
Conclusion
Previous, independent reports had suggested two distinct consequences of phosphorylating pleckstrin. Based on these reports we set out to further investigate the mechanism through which phosphorylation regulates pleckstrin with respect to its structure and function. The findings reported here demonstrate that pseudophosphorylation not only alters the oligomeric state of pleckstrin by significantly reducing dimerization but also causes a conformational change in the protein. Collectively these phosphorylation-dependent changes appear to regulate the function of pleckstrin by altering its phosphoinositide binding properties.
Acknowledgments
JIW was awarded a Canada Research Chair in Thrombosis and the Heart and Stroke Foundation of Ontario J.F. Mustard Chair in Cardiovascular Research. SGJ was a recipient of a Canadian Institutes of Health Research (CIHR) Canadian Graduate Scholarship. This work was supported in part by a Canadian Institutes of Health Research grant to MSJ (MOP 89903). This work was supported in part by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases (RG).
References
-
Lyons RM, Stanford N, Majerus PW (1975) Thrombin- Induced Protein Phosphorylation in Human Platelets. J Clin Invest 56(4): 924-936.
-
Lyons RM, Atherton RM (1979) Characterization of a Platelet Protein Phosphorylated during the Thrombin- Induced Release Reaction. Biochemistry 18(3): 544-552.
-
Tyers M, Haslam RJ, Rachubinski RA, Harley CB (1989) Molecular Analysis of Pleckstrin: The Major Protein Kinase C Substrate of Platelets. J Cell Biochem 40(2): 133-145.
-
Gailani D, Fisher TC, Mills DC, Macfarlane DE (1990) P47 Phosphoprotein of Blood Platelets (Pleckstrin) is a Major Target for Phorbol Ester-Induced Protein Phosphorylation in Intact Platelets, Granulocytes, Lymphocytes, Monocytes and Cultured Leukaemic Cells: Absence of P47 in Non-Haematopoietic Cells. Br J Haematol 74(2): 192-202.
-
Jezyk MR, Snyder JT, Gershberg S, Worthylake DK, Harden TK, et al. (2006) Crystal Structure ofRac1 Bound to its Effector Phospholipase C-beta2. Nat Struct Mol Biol 13(12): 1135-1140.
-
Worthylake DK, Rossman KL, Sondek J (2004) Crystal Structure of the DH/PH Fragment of Dbs without Bound GTPase. Structure 12(6): 1078-1086.
-
Lu M, Kinchen JM, Rossman KL, Grimsley C, deBakker C, et al. (2004) PH Domain of ELMO Functions in Trans to Regulate Rac Activation Via Dock180. Nat Struct Mol Biol 11(8): 756-762.
-
Abrams CS, Zhang J, Downes CP, Tang X, Zhao W, et al. (1996) Phosphopleckstrin Inhibits Gbetagamma- Activable Platelet Phosphatidylinositol-4,5- Bisphosphate 3-Kinase. J Biol Chem 271(41): 25192- 25197.
-
Edlich C, Stier G, Simon B, Sattler M, Goll CM (2005) Structure and Phosphatidylinositol-(3,4)- Bisphosphate Binding of the C-Terminal PH Domain of Human Pleckstrin. Structure 13(2): 277-286.
-
Harlan JE, Hajduk PJ, Yoon HS, Fesik SW (1994) Pleckstrin Homology Domains Bind to Phosphatidylinositol-4,5- Bisphosphate. Nature 371(6493): 168-170.
-
Baig A, Bao X, Wolf M, Haslam R (2009) The Platelet Protein Kinase C Substrate Pleckstrin Binds Directly to SDPR Protein. Platelets 20(7): 446-457.
-
Abrams CS, Wu H, Zhao W, Belmonte E, White D, et al. (1995) Pleckstrin Inhibits Phosphoinositide Hydrolysis Initiated by G-Protein-Coupled and Growth Factor Receptors. A Role for Pleckstrin’s PH Domains. J Biol Chem 270(24): 14485-14492.
-
Craig KL, Harley CB (1996) Phosphorylation of Human Pleckstrin on Ser-113 and Ser-117 by Protein Kinase C. Biochem J 314(pt 3): 937-942.
-
McDermott AM, Haslam RJ (1996) Chemical Cross- Linking of Pleckstrin in Human Platelets: Evidence for Oligomerization of the Protein and its Dissociation by Protein Kinase C. Biochem J 317 ( Pt 1): 119-124.
-
Ma AD, Brass LF, Abrams CS (1997) Pleckstrin Associates with Plasma Membranes and Induces the Formation of Membrane Projections: Requirements for Phosphorylation and the NH2-Terminal PH Domain. J Cell Biol 136(5): 1071- 1079.
-
Sloan DC, Wang P, Bao X, Haslam RJ (2002) Translocation of Pleckstrin Requires its Phosphorylation and Newly Formed Ligands. Biochem Biophys Res Commun 293(1): 640-646.
-
Lian L, Wang Y, Flick M, Choi J, Scott EW, et al. (2009) Loss of Pleckstrin Defines a Novel Pathway for PKC-Mediated Exocytosis. Blood 113(15): 3577-3584.
-
Roll RL, Bauman EM, Bennett JS, Abrams CS (2000) Phosphorylated Pleckstrin Induces Cell Spreading via an Integrin-Dependent Pathway. J Cell Biol 150(6): 1461- 1466.
-
Alim MA, Njenda D, Lundmark A, Kaminska M, Jansson L, et al. (2022) Pleckstrin Levels Are Increased in Patients with Chronic Periodontitis and Regulated via the MAP Kinase-p38 α Signaling Pathway in Gingival Fibroblasts. Frontiers in Immun 12: 801096.
-
Lundmark A, Davanian H, Bage T, Johannsen G, Koro C, et al. (2015) Transcriptome Analysis Reveals Mucin 4 to be Highly Associated with Periodontitis and Identifies Pleckstrin as a Link to Systemic Diseases. Sci Rep 5: 18475.
-
Gabbeta J, Yang X, Sun L, McLane MA, Niewiarowski S, et al. (1996) Abnormal Inside-Out Signal Transduction- Dependent Activation of Glycoprotein IIb-IIIa in a Patient with Impaired Pleckstrin Phosphorylation. Blood 87(4): 1368-1376.
-
Yang X, Sun L, Ghosh S, Rao AK (1996) Human Platelet Signaling Defect Characterized by Impaired Production of Inositol-1,4,5-Triphosphate and Phosphatidic Acid and Diminished Pleckstrin Phosphorylation: Evidence for Defective Phospholipase C Activation. Blood 88(5): 1676-1683.
-
Yang X, Sun L, Gabbeta J, Rao AK (1997) Platelet Activation with Combination of Ionophore A23187 and a Direct Protein Kinase C Activator Induces Normal Secretion in Patients with Impaired Receptor Mediated Secretion and Abnormal Signal Transduction. Thromb Res 88(3): 317-328.
-
Ding Y, Kantarci A, Badwey JA, Hasturk H, Malabanan A, et al. (2007) Phosphorylation of Pleckstrin Increases Proinflammatory Cytokine Secretion by Mononuclear Phagocytes in Diabetes Mellitus. J Immunol 179(1): 647- 654.
-
Brumbaugh J, Schleifenbaum A, Gasch A, Sattler M, Schultz C (2006) A Dual Parameter FRET Probe for Measuring PKC and PKA Activity in Living Cells. J Am Chem Soc 128(1): 24-25.
-
Brumbaugh J, Schleifenbaum A, Stier G, Sattler M, Schultz C (2006) Single- and Dual-Parameter FRET Kinase Probes Based on Pleckstrin. Nat Protoc 1(2): 1044-1055.
-
Brumell JH, Craig KL, Ferguson D, Tyers M, Grinstein S (1997) Phosphorylation and Subcellular Redistribution of Pleckstrin in Human Neutrophils. J Immunol 158(10): 4862-4871.
-
Brumell JH, Howard JC, Craig K, Grinstein S, Schreiber AD, et al. (1999) Expression of the Protein Kinase C Substrate Pleckstrin in Macrophages: Association with Phagosomal Membranes. J Immunol 163(6): 3388-3395.
-
Schleifenbaum A, Stier G, Gasch A, Sattler M, Schultz C (2004) Genetically Encoded FRET Probe for PKC Activity Based on Pleckstrin. J Am Chem Soc 126(38): 11786- 11787.
-
Cole JL, Lary JW, Moody TP, Laue TM (2008) Analytical Ultracentrifugation: Sedimentation Velocity and Sedimentation Equilibrium. Methods Cell Biol 84: 143- 179.
-
Schuck P (2000) Size-Distribution Analysis of Macromolecules by Sedimentation Velocity Ultracentrifugation and Lamm Equation Modeling. Biophys J 78(3): 1606-1619.
-
Schuck P (2003) On the Analysis of Protein Self- Association by Sedimentation Velocity Analytical Ultracentrifugation. Anal Biochem 320(1): 104-124.
-
Abrams CS, Zhao W, Belmonte E, Brass LF (1995) Protein Kinase C Regulates Pleckstrin by Phosphorylation of Sites Adjacent to the N-Terminal Pleckstrin Homology Domain. J Biol Chem 270(40): 23317-23321.
-
Abrams CS, Zhao W, Brass LF (1996) A Site of Interaction between Pleckstrin’s PH Domains and G Beta Gamma. Biochim Biophys Acta 1314(3): 233-238.
-
Vieira OV, Botelho RJ, Grinstein S (2002) Phagosome Maturation: Aging Gracefully. Biochem J 366(pt 3): 689- 704.
- Evaluation of Proximate and Mineral Compositions of Momordica charantia L. (Cucurbitaceae)
- Targeting Superbugs: Efficacy of Bacteriophage Therapy against Antibiotic-Resistant Pseudomonas Aeruginosa in Urinary Tract Infections
- Genetic Insights into Prepubertal Gynecomastia: A Comprehensive Analysis of a Rare 45,X[2]/ 46,X, + mar[28] Karyotype
- The Efficiency of Biological Treatment Plants in Some Private Hospitals in the City of Basra, Iraq
- Exploring the Combined Efficacy of Carvacrol and Friedelin against Multi-Drug Resistant Bacteria in Upper and Lower Respiratory Tract Infections
- Isolation, Identification and Comparative Analysis of Oral Microbial Communities in Smokers and Non-Smokers: A Scientific Investigation